Какво представляват наследствените човешки заболявания. Генетично заболяване
Това се пресича! Оригиналът е взет от eka_tyryshkina Грешките на природата: хора с редки болести.
Тук се разболях онзи ден, както винаги, тъй като отивахме само там, където трябваше да посетим, болен съм! Толи, болестта ми отговаря на планираността на събитието, а също и на нещо, но е добре, че не реагира на работата. Като цяло болестта не е лесна за мен)
И ето, че съм болен в късен час, след като вече е свършил цялата работа, препрочитам и разглеждам всички интересни уебсайтове, изведнъж не очаквах да разбера за най-редките язви на планетата и знаете толкова много интересно и шокиращо !!!
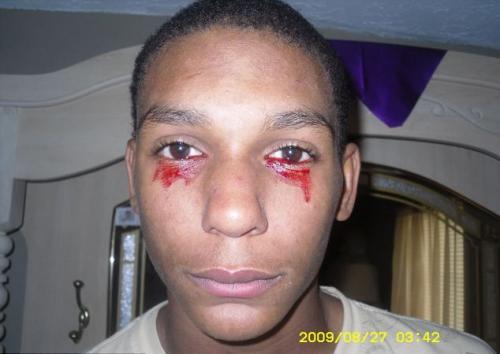
haemolacria
("Кървави сълзи") се наблюдава в един човек на милион.Кръвта, вместо сълза, изведнъж започва да тече от очите и това може да продължи около час. През деня пациентът излива кървави сълзи от 3 до 20 пъти.
Точната причина за това заболяване не е напълно изяснена и следователно не може да бъде лекувана. Медицинските специалисти досега са представили версията, че хемолакрията е едно от кръвните заболявания или тумори.
На снимката - 15-годишен Калвино Инман (Тенеси, САЩ)

Вампирен синдром
Диагностициран с "Вампирски синдром"
(ектодермална дисплазия) в света има само 7 хиляди души.
В допълнение към смъртоносната бледа кожа и остри кучешки зъби (при липса на част от зъбите), пациентите имат рядка и тънка коса, способността им да се потят се намалява, следователно тялото им е предразположено към прегряване. Симптомите се проявяват в детска възраст, но е възможно да се идентифицира заболяването още на етапа на бременността с помощта на генетични тестове.
Момчетата са принудени да носят слънчеви очила и да използват слънцезащитни продукти, когато излизат навън, тъй като не могат да бъдат под пряка слънчева светлина.В същото време физическото развитие и физическата активност остават нормални.Самата болест е нелечима, само симптомите могат да бъдат коригирани. По-специално, можете да възстановите нормалната форма на зъбите.
Болестта на Саймън е диагностицирана в ранна детска възраст. Когато Манди била бременна за втори път, тя била предупредена, че второто дете може да има същото заболяване.Въпреки това, Саймън се разви и се разви добре, така че родителите поеха този риск.
Момчетата казват: "Някои деца се смеят на нашия външен вид, но нашите приятели мислят, че е готино."
На снимката - Саймън (13-годишен) и Джордж (11-годишен) Кълън (Съфолк, Великобритания).

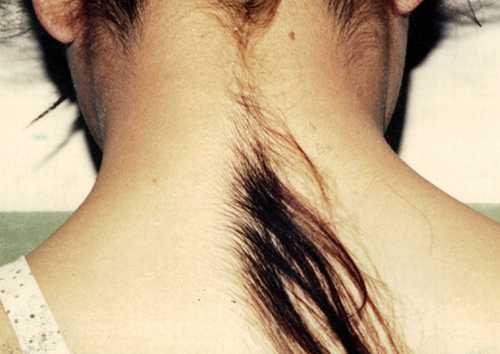
хипертрихоза
("Синдром на Върколака") е заболяване, което се проявява при прекомерен растеж на косата, което не е характерно за тази област на кожата, което не съответства на пола и възрастта. грозота ... Те подават заявления в Книгата на рекордите на Гинес - за да станат известни и да печелят пари ... Китайският Ю Дженхуанг успя да направи това за всички - благодарение на супер-космат, той основава най-популярната рок група в страната си и става милионер.
Не е известно защо се случва тази мутация. И никой все още не е разработил лечение за хипертрихоза. Козметиците могат да премахват косата само за достатъчно дълъг период ...
На снимката - 6-годишен Нат Сасуфан (Тайланд), 2007 г.

На снимката - 33-годишният Ю Дженхуанг (Китай), най-хубавият човек на света

Болест на слона
("Протеев синдром", elephantiasis, elephantiasis, elephantiasis) - увеличаване на размера на която и да е част на тялото, дължащо се на болезненото развитие на кожата и подкожната тъкан.
А най-известният пациент е „слонът“ - Джозеф Меррик. За известния британец през 1980 г. режисьорът Дейвид Линч дори прави филм, който е номиниран за "Оскар" в осем номинации ... Филмът е за човешкото достойнство ... на алкохолизираното тяло на Джозеф Мерик. Неговата подложка всеки ден взимаше актьора 12 часа на ден ...
На снимката - 35-годишен Mendi sellars (United Kingdom)

Аномалия на гена се състои в ускоряване стареене на тялото ,
- progeria - подразделени на детска стая (синдром на Гетчинсън) и възрастен (синдром на Вернер). За първи път за синдром на преждевременно стареене започнаха да говорят преди 100 години. И не е изненадващо, че такива случаи се срещат веднъж в 4-8 милиона бебета. Progeria (от гръцката Pro - преди, gerontos - Elder) е изключително рядко генетично заболяване, което ускорява процеса на стареене около 8-10 пъти. Казано по-просто, едно дете в една година расте с 10-15 години. Осемгодишният мъж изглежда 80 години - със суха набръчкана кожа, оплешивяваща глава ... Тези деца обикновено умират след 13-14 години след няколко сърдечни пристъпи и инсулти на фона на прогресивна атеросклероза, катаракта, глаукома, пълна загуба на зъби и др. И само няколко живеят до 20 години или повече.
Сега само 42 случая на прогерия хора са известни в света ... От тях, 14 души живеят в Съединените щати, 5 в Русия, останалите в Европа ...
В момента съществуват няколко организации, които предоставят помощ на малките стари хора и техните семейства. В интернет има сайтове, посветени на този конкретен проблем, някои от тях са отворени за лекари или социални работници, а други за семействата на пациентите.
На снимката - 24-годишният Леон Бот



38-годишен хората Tree
Деде Косвара, който живее на остров Ява, в Индонезия, стана известен по целия свят заради човешкия папиломен вирус, който обикновено води до появата на малки брадавици, но в случая с индонезийците той е деформирал крайниците му до непознаване.
Проблемът на Деде е, че той има рядко генетично заболяване, което не позволява на имунната му система да задържи растежа на тези брадавици. Следователно, вирусът е бил в състояние да "завладее клетъчния механизъм на нейните кожни клетки", като им дава заповеди да произвеждат голямо количество роговица, от която са били съставени. Dede също имаше нисък брой на белите кръвни клетки.


Заболяване с пеперуди
Хиперпластичната булева епидермолиза е генетично заболяване, което се проявява в първите дни от живота. Всъщност кожата на новороденото е толкова деликатна, че всяко докосване води до появата на рани и мехури. Най-засегнатите области са: лакти, колене, крака, ръце. Получаващата се язва, от която кожата се разтваря на слоеве, не лекува дълго време, освобождава течност от нея. След образуването на голям пурпурен белег.
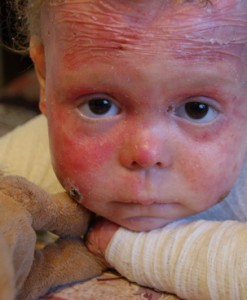
Няма лечение за това заболяване, възможно е само облекчаване на симптомите. Неотдавна историята на Лиза Кунигел, която живее с булева епидермолиза в продължение на почти десет години, разтърси цяла Русия. Няколко пъти на ден, тя се нуждае от превръзки и лечение с антимикробни мехлеми и гелове. В допълнение, всички 9 години Lisa съпътства болката.
Синдром на русалка
Една от най-редките аномалии в развитието е сиреномелия, наречена "синдром на русалка". При този дефект новородените се раждат с подредени крака, които приличат на рибена опашка. Те имат само един бъбрек, няма гениталии. Поради голямото увреждане на вътрешните органи, такива бебета обикновено умират скоро. Заболяването се среща при един от 100 000 новородени. През всичките години на наблюдение, само три бебета са успели да оцелеят. Един от тях беше Шило Пепин.
Шило е роден през 1999 г. и става най-известното дете със синдрома на "русалка". По време на десетте години, в които можеше да живее, тя имаше хиляди приятели по целия свят, които подкрепяха момичето и майка й. Shilo се опита да води пълен живот - тя, като всички обикновени деца, ходи на училище, посещаваше уроци по танци, ходи в увеселителни паркове. Момичето стана известно след участието си в шоуто на Опра Уинфри. Научавайки Chanel направи няколко филма за нея, стотици сайтове в интернет са посветени на нея.

Историята на Сило е удивителна история за чудо. Дете, което през цялото си детство се бореше да оцелее. Малко момиче, което можеше да се наслаждава на всеки ден, въпреки неизлечимата болест.
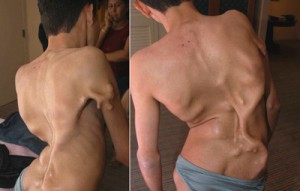
Болестта на Мунхаймер
Фибродиплазията е изключително рядко заболяване. Официалната статистика е следната: 1 пациент на 2,000,000 души. Болестта на Мунхаймер възниква в резултат на генна мутация и при раждането се проявява във външни дефекти. Бебето има усукани големи пръсти, гръбначен стълб. Патологията води до увреждане, ранна смъртност. Когато трябва да се проведат противовъзпалителни процеси, растежът на костите започва да се формира, поради което заболяването често се нарича "второ скелетно заболяване".
Всяко, дори и леко нараняване, може да доведе до развитие на остъкляване в засегнатата област.В момента няма официално лечение на смъртоносната болест. Учените са разработили лекарство, което теоретично може да се бори с болестта. Необходимите клинични проучвания обаче все още не са проведени. Уви, много е трудно да ги държите - в целия свят има не повече от 600 души с болестта на Мюнчамер.
феномен "Линии Блашко" характеризиращо се с наличието на странни ленти по цялото тяло. Линията Блашко е невидим модел, заложен в ДНК. И проявлението на болестта става появата на този модел.
Обикновено моделът на гърба е V-образен, а на гърдите, корема и страните - S-образен.
Причината за заболяването може да бъде мозаицизъм. Във всеки случай, появата на линиите на Блашко по никакъв начин не е свързана с човешката нервна, мускулна и лимфна системи.
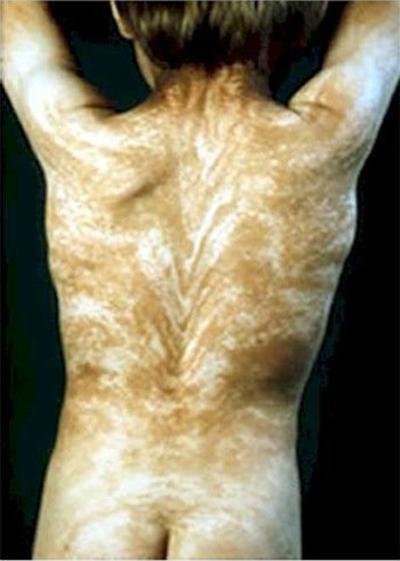
Друга анормална болест е акантозис нигрикансили синдром на синята кожа. Хората с тази диагноза могат да имат синя, индигова, слива или почти лилава кожа.В 60-те години на миналия век в Кентъки живее цялото семейство от „сини” хора. Бяха известни като Сините Фуге. Тази функция е предавана от поколение на поколение.


Около 6% от жителите на света страдат от редки болести и този брой продължава да нараства. Всички уникални болести имат различно естество, но по-голямата част от феноменалните заболявания са свързани с генетични аномалии и инфекции.
Могат да се наследяват не само външни признаци, но и болести. Прекъсванията в гените на предците водят, като резултат, до последствия в потомството. Ще говорим за седемте най-често срещани генетични заболявания.
Наследствените свойства се предават на потомци от техните предци под формата на гени, обединени в блокове, наречени хромозоми. Всички клетки на тялото, с изключение на пола, имат двойно множество хромозоми, половината от които идват от майката, а втората част идва от бащата. Заболяванията, причинени от някои генни неуспехи, са наследствени.
късогледство
Или късогледство. Генетично обусловено заболяване, чиято същност е, че образът се формира не върху ретината, а пред нея. Най-честата причина за това явление е увеличеното око. По правило миопията се развива по време на юношеството. Човекът в същото време вижда много добре отблизо, но не вижда добре в далечината.
Ако и двамата родители са недалновидни, тогава рискът от късогледство при децата им е над 50%. Ако и двамата родители имат нормално зрение, тогава вероятността от развитие на късогледство е не повече от 10%.
Изследвайки късогледството, служителите на Австралийския национален университет в Канбера заключиха, че 30% от бялата раса са присъщи на късогледството и засягат до 80% от родените в Азия хора, включително жители на Китай, Япония, Южна Корея и др. Учените са идентифицирали 24 гена, свързани с миопия, и също потвърждават връзката си с два предварително установени гена. Всички тези гени са отговорни за развитието на окото, неговата структура, предаване на сигнала в очната тъкан.
Синдром на Даун
Синдромът, кръстен на английския лекар Джон Даун, който го описа за пръв път през 1866 г., е форма на хромозомна мутация. Синдромът на Даун засяга всички раси.
Заболяването се дължи на факта, че в клетките не са два, а три случая на 21-та хромозома. Генетика го нарича тризомия. В повечето случаи допълнителната хромозома се предава на детето от майката. Смята се, че рискът от раждане на дете със синдрома на Даун зависи от възрастта на майката. Въпреки това, поради факта, че като цяло те най-често раждат в младостта, 80% от всички деца със синдром на Даун се раждат от жени на възраст под 30 години.
За разлика от гените, хромозомните аномалии са случайни нарушения. А в семейството може да има само един човек, страдащ от подобна болест. Тук се срещат и изключения: в 3-5% от случаите има по-редки - транслокационни форми на синдрома на Даун, когато детето има по-сложен набор от хромозоми. Този вариант на заболяването може да се повтори в няколко поколения от едно и също семейство.
Според информацията на благотворителната фондация "Даунсайд нагоре" всяка година в Русия се раждат около 2500 деца със синдром на Даун.
Синдром на Klinefelter
Друго хромозомно разстройство. Приблизително всеки 500 новородени момчета отговарят за една с тази патология. Синдромът на Klinefelter обикновено се появява след пубертета. Страдащи от този синдром, мъжете са безплодни. В допълнение, те се характеризират с гинекомастия - увеличаване на млечната жлеза с хипертрофия на жлезите и мастната тъкан.
Синдромът получи името си в чест на американския лекар Хари Клайнфелтер, който за първи път описа клиничната картина на патологията през 1942 година. Заедно с ендокринолог Фулър Олбрайт, той установява, че ако жените обикновено имат двойка полови хромозоми на ХХ век, а мъжете - XY, тогава при този синдром при мъжете има от един до три допълнителни Х хромозоми.
Цветна слепота
Или цветна слепота. Тя е наследствена, много по-рядко придобита. Тя се изразява в невъзможността да се разграничат един или няколко цвята.
Цветната слепота е свързана с Х-хромозомата и се предава от майката, собственик на „счупения” ген на сина си. Съответно до 8% от мъжете и не повече от 0,4% от жените страдат от цветна слепота. Факт е, че при мъжете „бракът” в единствената Х-хромозома не се компенсира, тъй като вторият Х-хромозома, за разлика от жените, не е такъв.
хемофилия
Друго заболяване, наследено от синове от майки. Историята на потомците на английската кралица Виктория от династията Уиндзор е добре известна. Нито тя, нито родителите й страдаха от това сериозно заболяване, свързано с нарушения на коагулацията. Предполага се, че генната мутация настъпва спонтанно, поради факта, че бащата на Виктория по време на зачеването й е вече на 52 години.
От Виктория "фаталният" ген е наследен от деца. Нейният син Леополд умира поради хемофилия на 30-годишна възраст, а две от петте й дъщери - Алис и Беатрис - са носители на злополучния ген. Един от най-известните потомци на Виктория, страдащ от хемофилия, е син на внучката си, Царевич Алексей, единственият син на последния руски император Николай II.
Кистозна фиброза
Наследствено заболяване, което се проявява в нарушаване на жлезите на външната секреция. Характеризира се с повишено изпотяване, секреция на слуз, която се натрупва в тялото и предпазва детето от развитие и, най-важното, предотвратява пълното функциониране на белите дробове. Вероятно е фатално поради дихателна недостатъчност.
Според руския клон на американската химико-фармацевтична корпорация Abbott, средната продължителност на живота на пациентите с муковисцидоза е 40 години в европейските страни, 48 години в Канада и САЩ и 30 години в Русия. От известните примери е полезно да се спомене френският певец Грегъри Лемархал, който почина на 23-годишна възраст. Предполага се, че кистозната фиброза и Фредерик Шопен, които са починали в резултат на неуспех на белите дробове на възраст от 39 години, страдат.
Заболяването, посочено в древния египетски папирус. Характерен симптом на мигрена е епизодично или редовно силно главоболие в половината на главата. Римският лекар от гръцки произход Гален, който е живял през II век, нарича болестта хемикраний, което се превежда като "половината от главата". От този термин идва думата "мигрена". През 90-те години. Двадесети век е установено, че мигрената се дължи главно на генетични фактори. Бяха открити редица гени, отговорни за наследяването на мигрена.
Не само външни черти и черти на характера могат да бъдат прехвърлени на детето от неговите биологични родители, но и редица здравни проблеми.
Наследствените заболявания са редки, но по правило това са доста сериозни заболявания, които на практика не подлежат на лечение.
Всеки ген на човешкото тяло съдържа уникална ДНК, има свой собствен уникален код на специфична черта.
В тази ситуация е необходимо да се потърси помощ от лекар - генетика, да се подложи на генетично консултиране, да се установи степента на риск от конкретно генетично заболяване.
Болест на Даун
Днес, едно от най-често срещаните заболявания

е наследствена, е болест на Даун. Статистиката показва, че това заболяване се среща при едно новородено от седемстотин бебета. Тази диагноза обикновено се установява от специалист, който все още се намира в родилния дом, за период от 3-5 дни от живота на новородено дете.
За да се потвърди тази диагноза, се провежда процедура като кариотипно изследване. Това е изследване на множеството хромозоми при новородено бебе. Детето, което е болно, има седем хромозоми, един повече от здрав човек. Това заболяване възниква както при момчета, така и при момичета, като в този случай сексът не играе никаква роля.
Болест на Шершевски-Търнър
Това заболяване е характерно само за женските деца. Първите признаци на тази генетична патология могат да бъдат открити на възраст 10-12 години.
Като правило, на гърба на главата косата расте много бавно и освен това те имат дълбоко вкоренен корен. На възраст от 15 до 16 години и дори по-възрастни, момичетата не съществуват и именно това е причината за търсене на специалист. С възрастта болестта може да предизвика проявление на някои проблеми с умственото развитие на детето. Генетичната структура на болестта на Шершевски-Търнър при момичетата се характеризира с липсата на една Х-хромозома.
Болест на Клайнфелтер
Болестта на Klinefelter е генетично заболяване, което се проявява само при момчета. Първите признаци на заболяването могат да бъдат открити, когато детето навърши 15-16 години.
Първите признаци са:

При изследване на хромозомите болестта на Klinefelter се характеризира с увеличен брой: още една Х хромозома. В някои случаи могат да присъстват и други хромозоми: Y, XX, XY.
Мултифакторни генетични заболявания
Мултифакторните генетични заболявания са генетични патологии, които могат да възникнат при новородено дете във всяко семейство.
В този случай причината за развитието на такива заболявания е не само генетичните аномалии, но и редица външни фактори, например лоша екология, нарушен ритъм на живота на родителите.
Тези заболявания включват: коронарна болест на сърцето, заболявания на стомаха, както и проблеми с кръвоносната система.
Вродени дефекти, които са свързани с мултифакторни генетични заболявания, са цепната устна, цепнато небце и спина бифида.
В момента всички многобройни патологии могат да бъдат идентифицирани с помощта на съвременна апаратура: ултразвуково изследване на плода ще може да разкрие много аномалии в развитието на детето.
Генетичните заболявания са редки и комплексни по характер, заболявания, които почти не се лекуват поради нарушения на геномното ниво. Ето защо експертите препоръчват при планиране на дете предварително да преминат консултация с генетик, за да се избегнат проблеми в бъдеще.
Експерти ще разкажат за генетичните заболявания на видео:
Забелязахте грешка? Изберете го и щракнете върху Ctrl + Enterда ни уведомите.
Харесва ли ви? Laykni и запишете на вашата страница!
Генетичните заболявания са уникални, тъй като не зависят от начина на живот на човека, не могат да бъдат осигурени от просто преставане да ядат мазни храни или да започват да тренират сутрин. Те произтичат от мутации и могат да бъдат предавани от поколение на поколение.
Рядко наследствено заболяване, при което човек умира от неспособност да спи. Досега тя се чества само в 40 семейства по целия свят. Смъртоносно безсъние обикновено настъпва между 30 и 60 години (най-често след 50 години) и продължава от 7 до 36 месеца. С напредването на заболяването пациентът страда от по-тежки нарушения на съня и никакви хапчета за сън не му помагат. На първия етап безсънието е съпроводено с пристъпи на паника и фобии, при втория етап се добавят халюцинации и повишено изпотяване. В третия етап на заболяването, човекът напълно губи способността си да спи и започва да изглежда много по-възрастен от годините си. След това се развива деменция и пациентът умира - обикновено от изтощение или пневмония.

Нарколепсийно-катаплексичният синдром, който се характеризира с внезапни пристъпи на сън и релаксация на мускулите на тялото, също има генетична природа и се причинява от разстройства на фазата на бърз сън. Това се случва много по-често от фаталното семейно безсъние: 40 на всеки 100 хиляди души, еднакво при мъжете и жените. Човек, страдащ от нарколепсия, може внезапно да заспи за няколко минути в средата на деня. “Спящите атаки” напомнят за REM съня и могат да се случват много често: до 100 пъти на ден, с или без главоболие, което ги предшества. Често те се провокират от бездействие, но могат да се появят в напълно неподходящо време: по време на полов акт, спортуване, шофиране. Човек се събужда отпочинал.
![]()
Синдромът на Yuner Tan (SYT) се характеризира предимно с факта, че хората, страдащи от него, ходят на четири крака. Той е открит от турски биолог Юнер Тан, след като е изучил пет члена на семейството на Улас в селска Турция. Най-често хората с ФТК използват примитивна реч и имат вродена церебрална недостатъчност. През 2006 г. за семейството на Ulas е заснет документален филм, озаглавен „Семейството, ходещо на четири крака“. Тан го описва по следния начин: „Генетичната природа на синдрома предполага обратен етап в еволюцията на човека, най-вероятно причинен от генетична мутация, обратната на прехода от квадропедализъм (ходене на четири крайника) към двуполярност (ходене по две). В този случай, синдромът съответства на теорията на периодичното равновесие.

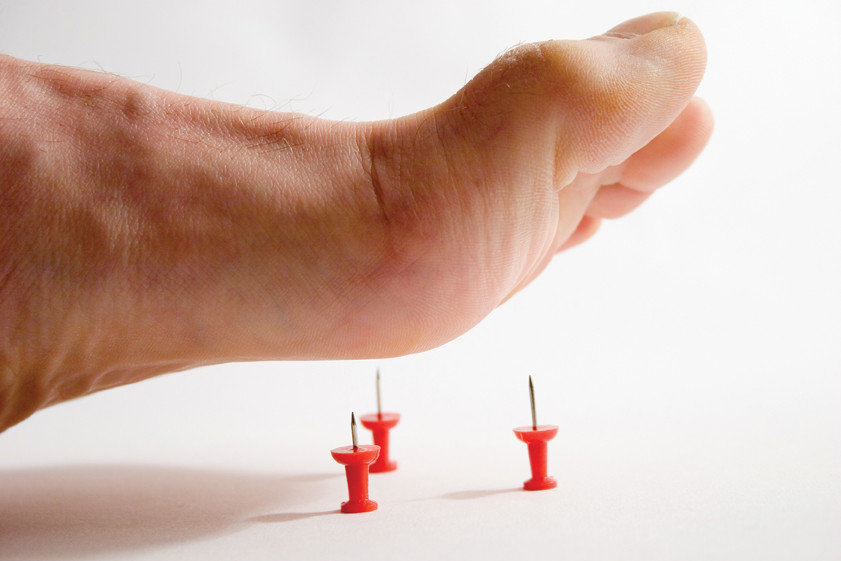
Една от най-редките заболявания в света: този тип невропатия се диагностицира при двама от един милион души. Аномалията е причинена от увреждане на периферната нервна система, което се дължи на прекомерното количество на гена PMP22. Основният признак за развитие на наследствената сензорна невропатия от първия тип е загубата на чувствителност на ръцете и краката. Човек престава да изпитва болка и чувства промяна в температурата, което може да доведе до некроза на тъканите, например, ако не разпознаете фрактура или друга вреда навреме. Болката е една от реакциите на тялото, която сигнализира за "неизправности", така че загубата на чувствителност към болка е изпълнена с твърде късно откриване на опасни заболявания, било то инфекции или язви.
Хората, страдащи от това необичайно заболяване, изглеждат много по-възрастни от възрастта си, поради което понякога се наричат \u200b\u200b"обратния синдром на Бенджамин Бътън". Поради наследствена генетична мутация, а понякога и в резултат на употребата на определени лекарства в организма, се нарушават автоимунните механизми, което води до бърза загуба на подкожни мастни резерви. Мастната тъкан на лицето, шията, горните крайници и тялото най-често страда, което води до бръчки и гънки. Досега са потвърдени само 200 случая на прогресивна липодистрофия и се развиват главно при жени. При лечението лекарите използват инсулин, „стягане” на лицето и инжекции с колаген, но това дава само временен ефект.

Хипертрихозата се нарича също „синдром на върколака” или „синдром на Абрамс”. Тя се проявява само в един човек от един милиард и само 50 случая от средновековието са документирани. Хората, страдащи от хипертрихоза, се характеризират с прекомерно количество коса по лицето, ушите и раменете. Това се дължи на разпадането на връзките между епидермиса и дермата по време на образуването на космените фоликули при тримесечен плод. Като правило, сигналите от получената дерма "информират" фоликулите за тяхната форма. На свой ред фоликулите също сигнализират на кожните слоеве, че вече има един фоликул в тази област и това води до факта, че космите по тялото растат приблизително на същото разстояние един от друг. В случай на хипертрихоза, тези връзки са счупени, което води до образуване на твърде гъста коса на тези части на тялото, където не трябва да бъде.

Ако някога сте чували за припадък от коза, тогава знаете как изглежда вродената миотония - поради мускулни спазми, човек замръзва за известно време. Причината за вродена (вродена) миотония е генетично заболяване: поради мутация функционирането на хлорните канали на скелетните мускули е нарушено. Мускулната тъкан е "объркана", произтичат контракции и релаксация, а патологията може да засегне мускулите на краката, ръцете, челюстите и диафрагмата.

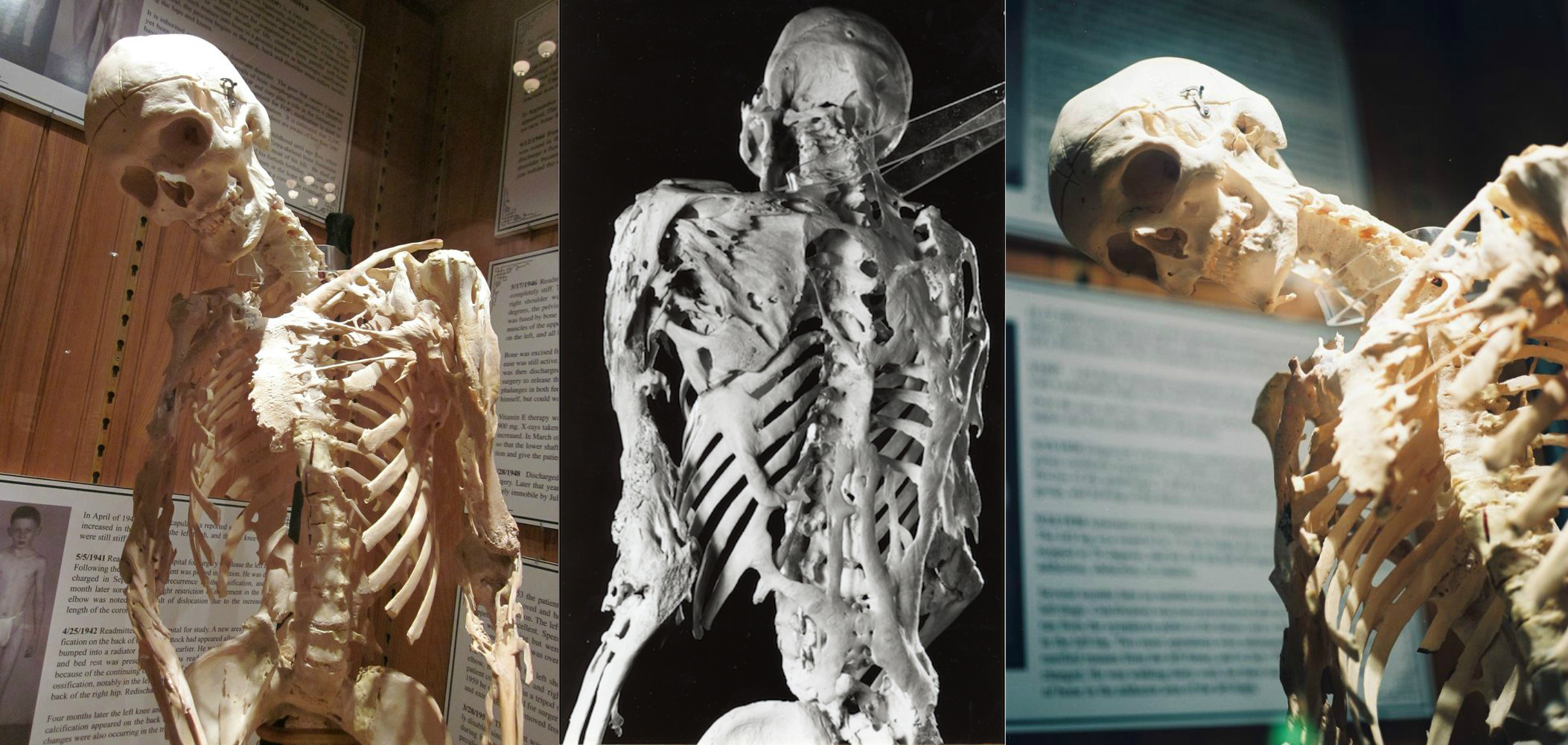
Рядко генетично заболяване, при което тялото започва да образува нови кости - осифицирани - в погрешни места: вътре в мускулите, сухожилията, сухожилията и други съединителни тъкани. Всяко нараняване може да доведе до образуването им: контузия, нарязване, фрактура, интрамускулна инжекция или хирургична намеса. Поради това е невъзможно да се премахнат осефиатите: след операцията костта може да стане по-силна. Физиологично, осификатите не се различават от обикновените кости и могат да издържат на значителни натоварвания, само че не са на правилното място.
FOP се причинява от мутация в ACVR1 / ALK2 гена, който кодира костен морфогенетичен протеинов рецептор. Тя се предава на човек по наследство от един от родителите, ако той също е болен. Да бъдеш носител на това заболяване е невъзможно: пациентът или е болен, или не. Досега FOP е сред нелечимите болести, но в момента се провежда втора поредица от изпитвания на лекарство, наречено паловаротен, което ви позволява да блокирате гена, отговорен за патологията.
Това наследствено заболяване на кожата се проявява в повишената чувствителност на човек към ултравиолетовите лъчи. Това се случва поради мутация на протеини, отговорни за възстановяване на увреждане на ДНК, което се проявява при излагане на ултравиолетова радиация. Първите симптоми обикновено се появяват в ранна детска възраст (до 3 години): когато детето е на слънце, той има сериозни изгаряния след няколко минути излагане на слънце. Също така, болестта се характеризира с появата на лунички, суха кожа и неравномерна промяна в цвета на кожата. Според статистиката, хората с пигментна ксеродерма са изложени на по-голям риск от развитие на рак: при липса на подходящи превантивни мерки около половината от децата, страдащи от ксеродерма, развиват определени видове рак на десетгодишна възраст. Има осем вида заболявания с различна тежест и симптоми. Според европейски и американски лекари болестта се среща в около четири милиона души.

Любопитно име за болестта, нали? Въпреки това, има един научен термин за този "болки" - десквамационен глосит. Географският език се проявява при около 2,58% от хората и най-често заболяването има хронични свойства и се влошава след хранене, по време на стрес или хормонален стрес. Симптомите се проявяват във външния вид на езика на избелени гладки петна, наподобяващи острови, тъй като болестта има такъв необичаен псевдоним, и с течение на времето някои от “островите” променят формата и местоположението си, в зависимост от това кои от вкусовите пъпки, разположени на езика, лекуват, и, напротив, са раздразнени.
Географският език на практика е безвреден, ако не вземете под внимание свръхчувствителност към пикантна храна или някакъв дискомфорт, който може да причини. Причините за това заболяване не са известни на медицината, но има доказателства за генетична предразположеност към неговото развитие.
Като рядко срещано заболяване за 2 милиона, синдромът на камъните, известен в медицината като прогресивна осифицираща фибродиплазия (FOP), е генетично заболяване, причинено от мутация в гените, която позволява на увредената съединителна тъкан да се превърне в кост. Хората с каменна болест създават нова структура на скелета. Прераждането започва с костите на шията и се разпространява надолу, засягайки всички структури, включително сакрума.
По-рано имаше няколко случая на това заболяване. Скелети, показващи осификация на съединителна тъкан, могат да бъдат намерени в Музея на медицинската история Mutter, разположен във Филаделфия, САЩ и в Лондон. Всъщност те са част от колекцията на музея Хънтър в Кралския колеж на хирурзите.
Синдромът на камъните започва да се появява в ранна възраст, през годините се развиват все повече нови кости. Повечето пациенти успяват да живеят около 40 години, тъй като след тази възраст вероятността от смърт се увеличава в резултат на проблеми с дишането. Спортните наранявания, паднали наранявания или дори инвазивни медицински процедури са опасни за такива хора, тъй като те могат да предизвикат интензивно възпаление на мускулите, сухожилията и сухожилията, вместо които, въпреки заздравяването, се образуват кости, които заменят увредената съединителна тъкан.
Причини за възникване на
Синдромът на камъните е автозомно доминантно генетично заболяване, причинено от мутация в ACVR 1 гена (активин-1 рецептор). В повечето случаи това разстройство е спонтанно и се появяват нови мутации при липса на фамилна анамнеза за заболяването. Те могат да се появят в резултат на излагане на йонизиращи лъчения, химикали, наркотици и инфекции. Този ефект може да настъпи в утробата или скоро след раждането. Въпреки това, няколко епизода от прехвърляне на гени от родител към потомство са записани в историята на случая. При такива обстоятелства, за проявлението на заболяването е достатъчен един променен алел (копие) на родителския ген.
При нормални условия генът ACVR 1 действа като модулатор, който контролира растежа и размножаването на мускулни клетки, връзки и други съединителни тъкани. Той кодира костен морфогенетичен протеин VMP 1, който контролира нормалната осификация и узряването на костите на скелета. При мутации, дължащи се на наранявания или вирусна инфекция в мускулите и съединителната тъкан, има постоянно активиране на гена с последващо освобождаване на дефектни протеини. Това води до отлагането на костни клетки в увредените участъци, прекомерния растеж на костите и сливането на ставите и костите.
симптоми
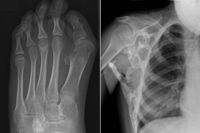
Симптомите на синдрома на камъните са доста различни и се развиват през целия живот. Те включват:
- Наличието на изкривени големи пръсти се счита за първи признак на това нарушение. Това също е важен симптом, тъй като помага на лекарите да го разграничат от други подобни състояния на опорно-двигателния апарат.
- Трудност при движение и скованост на ставите, в резултат на сливането им с новообразуваните кости.
- Трудност при хранене поради сливането на челюстните кости. В резултат на хранителни разстройства, обикновено има свързани заболявания. Сливането на челюстната става също води до проблеми с речта.
- С образуването на нови кости в гърдите, разширяването му е значително усложнено, което води до проблеми с дишането. Повечето пациенти със синдром на камъни умират от дихателна недостатъчност и пневмония.
- Хората, страдащи от това заболяване, имат характерен гръб на гърба, отклонение на гръбначния стълб встрани, затруднено движение и невъзможност за изпълнение на определени задачи. Тялото започва да изглежда като солидна статуя. Най-често през второто или третото десетилетие на живота пациентите стават напълно приковани.
Как да се справим
Към днешна дата не съществува лечение на синдрома на камъни. Въпреки това, използването на високи дози кортикостероиди може да помогне за намаляване на интензивността на възпалението на мускулната и съединителната тъкан, като по този начин забавя образуването на нови кости. Пациентите обикновено се съветват да избягват падания, наранявания и контактни спортове.
Перинатален тест за деца, родени с този заместник, не е рутинна процедура. Самата диагноза обаче може да бъде потвърдена чрез генетични изследвания.
Синдром на каменния човек в тийнейджърка

В продължение на много години Сини Немок, сладко 18-годишно момиче от Северен Кенсингтън (Лондон) се бори с рядко генетично заболяване. За първи път диагнозата "синдром на каменния човек" е направена от Сини на възраст от 12 години. В резултат на лек спад, възпалената област на гърба постепенно се замества с костни тумори и причинява мъчителна болка. Вратът и гръбначният стълб се сляха в едно, без да даде на момичето възможност да вдигне ръце над кръста. Сегашният й живот е засенчен от постоянни страхове от падания и наранявания, тъй като това със сигурност ще доведе до поникване на нови кости и ще влоши сегашното състояние.
Въпреки толкова трудна диагноза, Сини все още прекарва време с приятелите си. Обича да готви, да пазарува и, както всяко нормално момиче, обича грим. Нейното семейство не оставя надежда, че учените ще могат да разберат този сложен патологичен процес и да създадат лек за лечението на синдрома на каменния човек.
Сорокина Юлия Сергеевна
Как се проявява атопичен дерматит в детството? При повечето деца първите признаци на заболяването се появяват в ранна детска възраст. Техният вид обикновено се свързва с въвеждането на изкуствени смеси, краве мляко, яйца, риба и някои зърнени храни. Зачервяване, мехури, кожата става мокра, или, напротив, изсъхва и люспи по лицето, тялото, ръцете и краката на бебетата. Децата стават неспокойни, лошо спи. Ако не пропуснете момента и прибягвате до помощта на дерматолог, може да спрете алергичното възпаление на кожата. Възраст до 3 години е най-полезното време за лечение. В този период на детството, можете с максимална вероятност
При повечето деца първите признаци на заболяването се появяват в ранна детска възраст. Техният вид обикновено се свързва с въвеждането на изкуствени смеси, краве мляко, яйца, риба и някои зърнени храни. Зачервяване, мехури, кожата става мокра, или, напротив, изсъхва и люспи по лицето, тялото, ръцете и краката на бебетата. Децата стават неспокойни, лошо спи. Ако не пропуснете момента и прибягвате до помощта на дерматолог, може да спрете алергичното възпаление на кожата. Възраст до 3 години е най-полезното време за лечение. В този период на детството, можете с максимална вероятност
прекъсване на „марша“ на атопия.
На възраст 6-7 и 12-14 години е възможно обостряне на кожния процес. Провокира се от домашен прах, козина на домашни любимци, растителен прашец, бактерии, плесени и хранителни алергени, които отстъпват на заден план. Важна роля играят стресови ситуации, нарушаване на ежедневния режим, претоварване на детето с обучение, класове в много кръгове. Възпалението преминава в гънките на ръцете, краката, шията. Кожата става суха, удебелява се, образуват се корички на мястото на надраскване.
При някои хора проявите на атопия в ранна детска възраст са незначителни и остават без внимание на родителите и педиатрите, но в зряла възраст те се появяват отново, изглеждат “за първи път”. Възпалението на кожата възниква най-често в гънките на крайниците, по пръстите, в глезените, по лицето и шията. Обривът може да е влажен, екземен или сух. Комбинира техния тежък сърбеж и остатъчни ефекти под формата на удебелена груба кожа, пилинг, сухота.
Атопичен маршКато правило се развиват по-късно прояви на атопичен дерматит. През последните години говорим за така наречения "атопичен марш". Какво е това? Атопичният марш означава, че “диатезата” при децата може да послужи като начален етап за развитие на други, по-тежки форми на алергия - бронхиална астма, полиноза (алергия към полени), хранителни и лекарствени алергии, алергичен ринит (ринит) и конюнктивит (възпаление на очите) ), болки в ставите, мигрена. Освен това, тези заболявания могат да варират в различни периоди от живота в един човек. Например, имаше обрив по кожата, след това всичко си отиде и се появи полиноза или мигрена. Контролирайки хода на атопичния дерматит, лекарите и учените очакват да предотвратят "атопичния марш".
Атопия в наследство?Атопичният дерматит и атопията са вродени и наследствени състояния. Това означава, че има генетични фактори, които определят атопията. Тези механизми са внимателно проучени днес, за да служат за разработването на нови лечения за атопия в бъдеще.
Всички пътища на наследяване на атопичен дерматит все още не са напълно определени. Знаем, че ако един от родителите или най-близките роднини на детето има атопичен дерматит, астма или алергичен ринит, тогава детето ще развие атопичен дерматит с вероятност от 50%. В същото време, сред роднините на 30% от хората с атопичен дерматит, няма прояви на алергия.
Атопичната наследственост изобщо не е присъда, която осъжда човек през целия си живот или изпитва симптоми на алергия за известно време. Можете да бъдете "атопиком" и да нямате обрив и сърбеж.
Появата на човек с атопияКожата, страдаща от атопичен дерматит, е суха. Кожата на тялото и екстензорните повърхности на ръцете и / или краката са покрити с лъскави, тъмно оцветени малки „пъпки“. На страничните повърхности на раменете, лактите, понякога в областта на раменните стави може да има рогови гъсти малки папули ("ренде"). В по-възрастна възраст, кожата е разнообразна с наличието на тъмни и бели петна. Често пациентите в бузите са определени белезникави петна.
По време на ремисия единствените минимални прояви на атопичен дерматит могат да бъдат леко люспест, леко удебелена кожа или дори пукнатини в областта на прикрепването на ушната мида. В допълнение, такива признаци могат да бъдат хейлит (пукнатини, сухи устни), повтарящи се мехури (дълбоки пукнатини в ъглите на устата), средната пукнатина на долната устна и червена суха кожа на горните клепачи. Тъмните кръгове около очите, бледата кожа със земен нюанс могат да бъдат важни показатели за атопична предразположеност.
Как атопичният дерматит влияе на психологията?Психо-емоционалното влияние не се ограничава до случайни и краткотрайни преживявания поради обостряне на обрива. Неконтролираните прояви на атопичен дерматит постоянно усложняват живота на атопичния. Сърбежът причинява дискомфорт, а понякога води до безсъние, раздразнителност, депресия и повишена умора. Обривът на откритите части на тялото трябва да бъде скрит от другите, трябва винаги да помните, че нещо не може да се направи, или отново ще предизвика сърбеж и обрив.
Атопичният дерматит оказва силно въздействие върху цялостния начин на живот и външния вид на човека, понякога го прави наистина „различен”, гледайки по различен начин в един свят, в който има толкова много потенциално опасни алергични врагове, които другите обикновено търпят. Смята се, че "атопичният" по-предразположен към интелектуални занимания, внимателен и внимателен анализ на случващото се наоколо, по-чувствителен и затворен.
Проявите на атопичен дерматит засягат не само психологията на пациента, но и цялото семейство, хората около него и техните взаимоотношения.
Какво да правя atopiku? Много от нас са "атопични", 1-3% от всички възрастни европейци и 10-20% от учениците. Така се оказва: „това е нещо ежедневно!” Можем да преодолеем стреса и атопичния дерматит.
Много от нас са "атопични", 1-3% от всички възрастни европейци и 10-20% от учениците. Така се оказва: „това е нещо ежедневно!” Можем да преодолеем стреса и атопичния дерматит.
За да преодолеете емоционалния стрес и социалните проблеми, имате нужда от самочувствие и способност да контролирате състоянието на кожата си. Имате нужда от открита, поверителна дискусия за вашето състояние с приятели, роднини, лекари, други хора със същия проблем.
За да се поддържа състоянието на ремисия изисква специална грижа за кожата, дерматолозите са разработили специални инструменти - омекотители. Много известни марки на козметика имат линия от продукти за атопично
Кожа: Aven, La Rosh Pose и др. С постоянното използване на перилни гелове и кремове за тяло от тази серия възстановяваме защитната бариера на кожата и премахваме неадекватните реакции.
Какво друго можете да използвате за атопия? Разбира се, сорбенти! Това са вещества, които се поглъщат, свързват токсините, различните съединения и ги отстраняват от тялото по естествен път. като един от сорбенти, които познавам от дълго време. Сорбенти са показани за лечение на атопичен дерматит. Малките пациенти с "атопично" заболяване е трудно да изпият чаша вода с хапчета със сорбент в една глътка. Финият прах е най-подходящ за деца, лесно се разделя на малко количество вода, за разлика от грубите частици, което улеснява неговото приемане. След това можете да пиете чаша вода за половин час. След двуседмичен курс, често се появяват признаци на обостряне, което е преминало, дори и без използването на специфично лечение. Възрастен атопичен сорбент се приема както по време на острата фаза на дерматит, така и за профилактика преди сезонно обостряне.
Къде трябва да се лекува пациент с атопичен дерматит?Необходимо е да бъде наблюдаван и лекуван от същия дерматолог. При необходимост дерматологът предписва консултации на медицински специалисти: невролог, гастроентеролог, алерголог, вертебролог и др. Единственият начин да се осигури приемственост на различните видове терапия е да се проследи реакцията на пациента към тях.
За съжаление, ние трябва да се справим с пациенти, които, без да са завършили предписания курс на лечение, търсят нововъзникнали външни лекарства, преминават от един лекар на друг и всеки път, без да имат очакван ефект, изпитват допълнителен стрес, последвано от обостряне на заболяването. Не се лекувайте самостоятелно. Това е сериозно заболяване, което може да доведе до опасни усложнения и, както беше казано, до развитието на други форми на "атопия". За да контролирате болестта, предотвратете усложнения, трябва да следвате препоръките на лекуващия лекар дерматолог.
Най-редките болести в света
Както примитивните пещерни хора, така и съвременните брилянтни учени - човечеството винаги се е борило и се бори с много болести. Има група наследствени заболявания, които е трудно да се избегнат, ако родителите ви са болни; Някои болести са продукт на непредвидими генетични мутации. Голяма група заболявания е резултат от възпроизводството и просперитета на микроскопичните организми, които са се заселили в човешкото тяло.
Синдром на Гилбърт
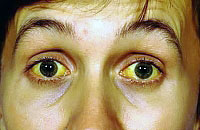
![]() Синдромът на Gilbert (SJ) е генетично заболяване и води до нарушаване обмена на билирубин, което може да предизвика доброкачествена неконюгирана хипербилирубинемия. Метаболизмът на билирубин е нарушен поради липса на глюкуронилтрансфераза, специален чернодробен ензим. Това предизвиква повишаване на нивото на несвързания билирубин в кръвта и появата на жълтеница. По синдром на Гилбърт, като правило, няма заплаха за плода. Присъствието на SJ увеличава риска от развитие на жлъчнокаменна болест.
Синдромът на Gilbert (SJ) е генетично заболяване и води до нарушаване обмена на билирубин, което може да предизвика доброкачествена неконюгирана хипербилирубинемия. Метаболизмът на билирубин е нарушен поради липса на глюкуронилтрансфераза, специален чернодробен ензим. Това предизвиква повишаване на нивото на несвързания билирубин в кръвта и появата на жълтеница. По синдром на Гилбърт, като правило, няма заплаха за плода. Присъствието на SJ увеличава риска от развитие на жлъчнокаменна болест.
Симптомите на синдрома на Гилбърт се проявяват по-ясно под въздействието на стрес, физическо натоварване, недохранване, след вирусни заболявания и в резултат на алкохол и някои лекарства. За болестта на Гилбърт се характеризират с:
- астения;
- пожълтяване на склерата и лигавиците на различна степен (жълтеността на кожата не винаги се наблюдава);
- болка в черния дроб;
- повишени нива на билирубин в кръвта;
- неизправност на стомаха и болезнено храносмилане.
Синдромът на Гилбърт е вроден, в който случай симптомите се появяват на възраст между 12 и 30 години. Вторият тип синдром е постхепатитен хипербилирубинемия, която се проявява след вирусен хепатит. Във втория случай е необходимо да се проведе диференциална диагноза, за да не се обърка LF с хроничен хепатит.
Диагностика на синдрома на Гилбърт
За диагностични изследвания и планиране на лечението, трябва да се свържете с терапевт, генетик, хематолог и гастроентеролог (хепатолог). Ако се подозира синдромът на Gilbert, в допълнение към анамнезата и физическия преглед се предписват следните диагностични методи:
- - При LF се наблюдава повишаване на хемоглобина (\u003e 160 g / l), ретикулоза и намаляване на осмотичната резистентност на еритроцитите.
- Биохимичният анализ на кръвта - билирубин може да достигне 6 mg / dL, но най-вече не надвишава границата от 3 mg / dL. Параметрите, които определят функцията на черния дроб, остават нормални. Алкалната фосфатаза може да се увеличи.
- PCR - генетичен маркер на SJ - броят на TA-повторите в промоторната верига на UGT1A1 гена.
- Ултразвуково изследване на жлъчния мехур и дуоденална интубация - почти всички пациенти със SJ имат промени в биохимичния състав на жлъчката.
- Чернодробна биопсия - възможни патологични промени в организма.
- Проба с гладно - в присъствието на SJ недохранване води до увеличаване на серумния билирубин.
- Тест с фенобарбитал - използването на фенобарбитал на фона на LF допринася за понижаване на нивото на неконюгиран билирубин.
- Проба с никотинова киселина в LF провокира увеличаване на съдържанието на неконюгиран билирубин. Същата реакция се появява, когато се прилага рифампицин.
Лекарят може да предпише допълнителни проучвания и диференциална диагноза на LF с друга хипербилирубинемия.
 Прогнозата е доста благоприятна, поради факта, че болестта на Гилбърт е сравнително безопасна и не се изисква специално лечение (това е по-скоро домашен характер). Основата на терапията е спазването на нормална диета, работа и почивка. По време на екзацербации, трябва да следвате диета номер 5 (отказ от мазни и пържени храни, алкохол), да вземат витамини и choleretic агенти. Терапевтът може да предпише курс на хепатопротектори. Важно е да запомните, че не трябва да прибягвате до топла физиотерапия. Лечението на SJ е насочено към възстановяване на нормалното ниво на уридин дифосфат глюкуронилтрансфераза (чернодробни ензими) и стабилизиране на общото състояние на пациента.
Прогнозата е доста благоприятна, поради факта, че болестта на Гилбърт е сравнително безопасна и не се изисква специално лечение (това е по-скоро домашен характер). Основата на терапията е спазването на нормална диета, работа и почивка. По време на екзацербации, трябва да следвате диета номер 5 (отказ от мазни и пържени храни, алкохол), да вземат витамини и choleretic агенти. Терапевтът може да предпише курс на хепатопротектори. Важно е да запомните, че не трябва да прибягвате до топла физиотерапия. Лечението на SJ е насочено към възстановяване на нормалното ниво на уридин дифосфат глюкуронилтрансфераза (чернодробни ензими) и стабилизиране на общото състояние на пациента.
Необходимо е да се консултирате с лекар и хепатолог, кои лекарства могат да се използват на фона на синдрома на Гилбърт и кои от тях трябва да бъдат изоставени (например, анаболни стероиди, глюкокортикоиди, кофеин и парацетамол могат да увеличат проявата на жълтеница).
Рак на червата: Симптоми и рискови фактори
 Рак на червата е злокачествен тумор, който се образува на лигавиците в различни части на червата. Най-често туморът може да се развие от полипи, но не всички се превръщат в онкология.
Рак на червата е злокачествен тумор, който се образува на лигавиците в различни части на червата. Най-често туморът може да се развие от полипи, но не всички се превръщат в онкология.
Рак на червата, тъй като той се развива, може да наруши нормалното функциониране на органа и да предизвика кървене, включително видими във фекални маси. Ако късното диагностициране на рак на червата се дължи на симптоми, то може да се разпространи и в други органи, което значително ще влоши прогнозата за лечението.
Рак на червата: Рискови фактори
Официалната медицина не посочва точните причини за рак на дебелото черво, но идентифицира основните рискови фактори, които увеличават вероятността от развитие на рак в червата:
- възраст: най-често хората с рак на червата са болни след 50 години;
- нездравословен начин на живот: ниска физическа активност, нездравословна храна, наднормено тегло, злоупотреба с алкохол и пушене;
- : възпалителни процеси в червата (болест на Крон, улцерозен колит и др.) се считат за предракови заболявания, провокиращи развитието на онкологията;
- наследственост: рискът от рак се увеличава, ако непосредствените роднини страдат от тях или други чревни заболявания.
Рак на червата: Симптоми
 В началните етапи на рак на червата, симптомите може да не привличат вниманието на пациента. Промените в честотата на движението на червата (в посока на намаляване или увеличаване), неразумната загуба на тегло, умората и слабостта, анемията и болката в ануса са основните признаци, които характеризират началото на онкологичния процес в червата.
В началните етапи на рак на червата, симптомите може да не привличат вниманието на пациента. Промените в честотата на движението на червата (в посока на намаляване или увеличаване), неразумната загуба на тегло, умората и слабостта, анемията и болката в ануса са основните признаци, които характеризират началото на онкологичния процес в червата.
С развитието на заболяването може да се появи постоянна болка в корема, подуване на корема и влошаване на общото благосъстояние. Появата на някой от симптомите трябва да бъде добра причина за незабавни посещения на лекар.
Когато ракът е открит на ранен етап, прогнозата за лечение е доста оптимистична - около 90% от пациентите остават живи и вече не отиват при лекаря да се оплакват. Въпреки това, повечето хора пропускат първите симптоми, приемайки ги за нарушения или хемороидални процеси, започват болестта, а след това дори операцията може да спаси живота само на около 60% от пациентите.
Защо се нуждаем от допълнителни зъби?
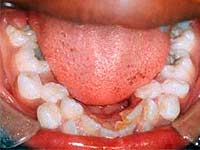 Допълнителните зъби, или хипердонтията, са една от наследствено определените аномалии на броя на зъбите, които са доста често срещани в момента. Около 2-3% от пациентите с малформации на зъбната система имат, в допълнение към 20 мляко или 32 постоянни зъби. Естеството на тази патология не е напълно ясно, смята се, че неговото появяване е свързано с нарушение на полагането на зъбите, или по-скоро, с нарушение на механизма на разделяне на зъбната пластина, което води до по-голям брой зъбни зачатъци.
Допълнителните зъби, или хипердонтията, са една от наследствено определените аномалии на броя на зъбите, които са доста често срещани в момента. Около 2-3% от пациентите с малформации на зъбната система имат, в допълнение към 20 мляко или 32 постоянни зъби. Естеството на тази патология не е напълно ясно, смята се, че неговото появяване е свързано с нарушение на полагането на зъбите, или по-скоро, с нарушение на механизма на разделяне на зъбната пластина, което води до по-голям брой зъбни зачатъци.
Къде да търсим допълнителни зъби?
Свръхдопълващи се зъби могат да бъдат открити при деца на ухапване от мляко, но по-често се откриват след смяна на зъбите при постоянен ухапване.
Обикновено около средните горни резци, молари, премолари, кучешки зъби се появяват допълнителни зъби, по-рядко между долните резци, премолари и кучешки зъби. Те могат да растат на зъбната дъга и могат да бъдат разположени на прага на устната кухина или директно в устната кухина в горната част на небцето.
Формата на извънбюджетните зъби може да бъде подобна на обичайните постоянни единици на зъбите, но по-често те са с форма на капка или спинообразна форма. Те могат да бъдат разположени отделно, споени с постоянни зъби, образуват цели зъбни конгломерати и зъбообразни образувания.
Понякога допълващите се зъби са „скрити” от очите, т.е. те са засегнати, тогава те се откриват само чрез рентгеново изследване.
Какви са опасните допълнителни зъби?
Устройствата с прекомерно допълване пречат на образуването на зъби и възпрепятстват изригването на постоянни зъби. При голям размер на челюстта един допълнителен зъб не нарушава структурата на зъбите, а с малка челюст със сигурност става причина за аномалии в положението на пълните зъби, което има отрицателни естетически, функционални и често психологически последици за човека.
При деца с необичайно стоматологично развитие често апетитът се намалява, по-бавно се дъвче храната, често се нарушава поглъщането и всичко това води до развитие на заболявания на храносмилателната система.
Близкото подреждане на зъбите, тяхното неправилно положение затруднява процесите на самопочистване на зъбите и провеждането на хигиенни процедури. Това създава благоприятни условия за развитие на кариес, гингивит, пародонтит и пародонтоза, водещи до разрушаване на твърдите тъкани на зъбите и загубата им.
Собствениците на допълнителни зъби често обикалят. Говорни нарушения и козметични дефекти причиняват присмех на детето, формират неинструментална и затворена личност от него и често засягат неговото умствено развитие.
Хипердонтична диагностика
В повечето случаи по време на изригването на предните зъби се откриват излишни зъби. За да се изясни техният брой и местоположение, е необходима рентгенова диагностика, но е необходим прост рентгенов анализ, тъй като сенките от допълнителните зъби се наслагват върху контурите на пълните зъби.
За точна диагностика на хипердонтичните рентгенови лъчи се използват вътре в устата с изображения в различни проекции. В случай на множествено задържане на свръхплъстени зъби, ортопантомографията дава полезна информация за взаимното подреждане на свръхпълните и постоянните зъби.
Какво да правим с допълнителни зъби?
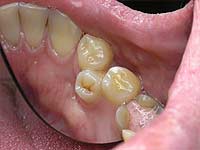 Подходът към лечението на хипердонтията е диференциран, терапията зависи от местоположението на допълнителните зъби. Като цяло, извънбюджетните зъби трябва да бъдат отстранени възможно най-рано, особено ако нарушават анатомията на зъбите, причинявайки болезнени преживявания у пациента. При елиминирането на допълнителни зъби в детството, нормалната форма на зъбите често се възстановява поради механизмите на саморегулиране на организма, но ако пропуснете времето, не можете да го направите без последващо ортодонтско лечение.
Подходът към лечението на хипердонтията е диференциран, терапията зависи от местоположението на допълнителните зъби. Като цяло, извънбюджетните зъби трябва да бъдат отстранени възможно най-рано, особено ако нарушават анатомията на зъбите, причинявайки болезнени преживявания у пациента. При елиминирането на допълнителни зъби в детството, нормалната форма на зъбите често се възстановява поради механизмите на саморегулиране на организма, но ако пропуснете времето, не можете да го направите без последващо ортодонтско лечение.
Понякога, за да се запазят функциите на зъбите, напротив, те жертват дистопичен постоянен зъб, като същевременно поддържат изгодно позициониран, анатомично завършен, пълен допълващ се зъб.
Ако на мястото на непроходим постоянен зъб расте, се определя степента на неговата полезност. Ако един излишен зъб е стабилен, има развит корен и е повече или по-малко анатомично правилна форма, и непроницаемият "законен собственик на мястото" е несъстоятелен и безполезен, предпочитание се дава на "нашественика".
Отстраняването на ударените свръхкомплексни зъби, потопени в челюстната тъкан, създава известни затруднения, свързани с тяхната дълбочина, неправилна форма, близост до корените и рудименти на постоянни зъби. Въпреки това, рационален оперативен подход към един излишен зъб, като се вземат предвид резултатите от рентгеновото изследване, позволява успешно решаване на тези проблеми.
При деца, извънбюджетните зъби се отстраняват под обща или локална анестезия, на фона на действието на успокоителни, при възрастни, локална анестезия е достатъчна.
Порфирия - научно обоснован "вампиризъм"
Вампиризмът е модерна субкултура, която обединява младите хора, които се смятат за вампири. По принцип, интересът се ограничава до изучаването на вампирските теми в изкуството и имитация на появата на любими герои, младите хора едва ли ще мислят за историята на произхода на образите на вампирите.
Случаи на "ненаучен" вампиризъм
Движението “вампиризъм” е създадено през 1970 г. благодарение на феновете на Ан Райс, автор на известния роман “Интервю с вампира”. Темата за вампирите обаче се корени в далечното минало и се отразява в фолклора на много народи.
Образът на вампира в изкуството еволюира през годините, но най-ярко отразени в готическия роман, публикуван от ирландския писател Дракула Брам Стокър, публикуван през 1897 г., последващите произведения дължат своето съществуване на това безсмъртно творчество.
Какви са литературните знаци на вампиризма?
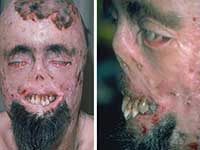 Най-често вампирите се изобразяват като интелигентни, елегантни, загадъчни и секси индивиди, водещи усамотен живот.
Най-често вампирите се изобразяват като интелигентни, елегантни, загадъчни и секси индивиди, водещи усамотен живот.
Те се нуждаят от кръв, за да поддържат метаболизма си и да не умират. Според легендите:
- вампирите се страхуват от слънчева светлина, защитени от тъмни дрехи, излизат само под нощ и се връщат преди зазоряване;
- дневната светлина убива вампир и намалява неговата сила;
- избягват вечери и вечери, човешката храна им е чужда;
- те са бледи, кожата е тънка и уязвима, студена на допир;
- непроменени - зъби и венци, докоснати от лилаво;
- очите на вампир са заобиколени от мъгла от пухкави мигли, катериците са червеникави и зениците са замъглени;
- те се характеризират с безпокойство, подозрителност, агресивност, насилие по време на остро желание за кръв, могат да се превърнат в чудовища във физически и психологически смисъл.
порфирия
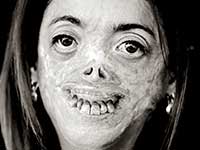 Без да притежават знания в областта на медицината, черпейки вдъхновение от ирландските митове за вампири, в традициите на народа на Трансилвания, историческите описания на живота на Влад Цепеш, прототипът на Дракула, Стокър, без да го знае, описва страданието на тежко болен човек с порфирия.
Без да притежават знания в областта на медицината, черпейки вдъхновение от ирландските митове за вампири, в традициите на народа на Трансилвания, историческите описания на живота на Влад Цепеш, прототипът на Дракула, Стокър, без да го знае, описва страданието на тежко болен човек с порфирия.
Порфирия, иначе - пурпурна болест, група заболявания, свързани с нарушен метаболизъм на порфирина, яркочервен пигмент. В основата на патологията са нарушения на синтеза на хем - съединение на порфирин с желязо, на базата на човешки еритроцити. Неуспехът в образователната система на heme води до анемия, натрупването в организма на продукти на междинния метаболизъм, които имат токсичен ефект върху органите и системите, които причиняват типичните симптоми на вампирска болест.
Причините за порфирията са генетични, болестта се наследява, Вероятността за предаване на порфириен ген е доста висока, болният родител “дава” дефектния ген на детето в 50% от случаите, независимо от пола, но само в 20% от случаите клиничната картина на заболяването се развива. Неговото проявление изисква действие. провокиращи факторинякои лекарства, инфекции, хормонални пренареждания, определени храни и алкохол - не за нищо, че митичните вампири избягват човешки празници.
Симптоми на порфирия
Тъй като болестите са по-чести сред мъжете и се проявяват през пролетните и летните месеци. Типичен признак на порфирия е урината червеникавокафява, поради наличието на оксидиран порфириноген, който се превръща в лилав порфирин.
Остра порфирия проява на силна болка в коремната област, долната част на гърба, крайниците, тахикардия, повишено кръвно налягане, повръщане, мускулна слабост, психомоторно възбуда, халюцинации, заблуди, епилептиформни припадъци и други симптоми, които се развиват в резултат на остро отравяне на тялото в резултат на метаболизъм на порфирина и дифузни периферни лезии \\ t и централната нервна система.
Порфирия има много типичен вид.
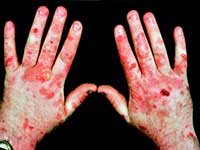
Можете да си представите реакцията на хората през Средновековието, когато са били изправени пред болна порфирия. Такъв спектакъл остави следа в паметта, запечатана в легендите, обрасли с митични подробности.
Порфирията е сериозно заболяване, основно лекувано с инфузия на хема. Освен това се предписват симптоматични средства, плазмафереза, кръвта е необходима на пациентите, за да се справят с интоксикацията и просто да живеят.
Истинският проблем за човека: безплодие и неговите причини
 Казват, че всеки човек в живота си трябва да посади дърво, да построи къща и да отгледа син. И ако практически всеки член на по-силния пол се справи с първите две задачи, ако желае, тогава, за съжаление, при решаването на последната от тях, до 8% от мъжете на Земята срещат сериозни проблеми. И тази цифра не взема предвид тези, които са се примирили с безплодието и са избрали да не споделят проблема си с лекарите.
Казват, че всеки човек в живота си трябва да посади дърво, да построи къща и да отгледа син. И ако практически всеки член на по-силния пол се справи с първите две задачи, ако желае, тогава, за съжаление, при решаването на последната от тях, до 8% от мъжете на Земята срещат сериозни проблеми. И тази цифра не взема предвид тези, които са се примирили с безплодието и са избрали да не споделят проблема си с лекарите.
Безплодието при мъжа е причина за бездетността на двойката
Безплодието не е просто отсъствие на деца, а диагностицирано е в случай, че двойката, която активно живее сексуално, без да използва контрацепция, среща трудности при зачеването в продължение на една година. Нека малко да изясним понятието "активен сексуален живот", за един е дневен секс, а за другия - няколко пъти месечно. В случай на безплодие - „активно“ означава поне 1 път седмично.
Според статистиката, в 40% от случаите на отсъствие на деца, двойката е виновна точно. Причините за това могат да бъдат различни, привидно безвредни и лесно преодолими, или толкова тежки, че е невъзможно да бъдат отстранени.
Мъжкото безплодие, както и жените, могат да бъдат абсолютни и относителни. Човек ще трябва да живее с абсолютно безплодие, ако има отстранени семенните жлези. Други опции за безплодие са потенциално относителни, изискват задълбочена диагноза, за да откриете причината и да разрешите проблема.
Варианти на мъжкото безплодие: причини и форми
 Процесът на образуване на пълноценни сперматозоиди е доста сложен и се контролира от хормон-произвеждащата част на мозъка - хипофизално-хипоталамусната система. Половите клетки, които се образуват в тестиса, узряват по пътя към семенните мехурчета, но липсата на проблеми в тази част от производството не гарантира успешното оплождане. Репродуктивната способност на човека се влияе от състоянието на всички мъжки органи и целия организъм.
Процесът на образуване на пълноценни сперматозоиди е доста сложен и се контролира от хормон-произвеждащата част на мозъка - хипофизално-хипоталамусната система. Половите клетки, които се образуват в тестиса, узряват по пътя към семенните мехурчета, но липсата на проблеми в тази част от производството не гарантира успешното оплождане. Репродуктивната способност на човека се влияе от състоянието на всички мъжки органи и целия организъм.
Какво причинява този проблем?
- Хипоталамо-хипофизни разстройства, които се срещат в мозъка, на нивото на хормонална регулация на сперматогенезата.
- Тестикулар, свързан с тестисите.
- Пост-тестикуларен, настъпващ по посока на сперматозоидите от тестиса, за да излезе от уретрата.
- Имунологични с наличието на антитела на сперматозоиди, които убиват сперматозоидите.
- Еякулаторни нарушения, т.е. проблемът с еякулацията.
- Сексуални разстройства, които нарушават доставката на сперматозоиди до женските полови органи, като еректилна дисфункция и намалено сексуално желание.
Способността на един човек да продължи състезанието се влияе неблагоприятно от:
- неблагоприятна екологична ситуация;
- стрес;
- медикаменти във високи дози;
- тютюнопушенето;
- прием на алкохол;
- употребата на наркотици.
Безплодие на тестисите
Безплодието на тестисите се появява при тестикуларно заболяване и неговата инертност.
- Разширени вени на скротума и семенната връзка (варикоцеле).
- Крипторхизъм, двустранен, не елиминиран или елиминиран сравнително късно, когато тъканта, продуцираща семена от тестисите, вече е атрофирана.
- Усукване на тестиса, водещо до драматично нарушаване на функцията му.
- Орхит или възпаление на тестиса, в който умират предшественици на сперматозоидите.
- Хипергонадотропен хипогонадизъм - тестикуларна хипоплазия.
- Генетични причини за нарушаване на формирането на репродуктивната система и формиране на тестисите.
- Резистентност към андрогени, когато сперматогенният епител на тестисите е глух до хормонални сигнали, провъзгласявайки образуването на зародишни клетки.
- Необструктивна азооспермия, състояние, при което не се нарушава производството на сперматозоиди, движението им по семепровода не е трудно, но изходът не разкрива "нито един" жив екземпляр, способен да продължи рода.
Посттестикуларни причини за безплодие
Тези причини водят до нарушаване на узряването на сперматозоидите, тяхната смърт, отслабване на способността им за торене и също така затрудняват движението на сперматозоидите по семепровода.
- Инфекции на гениталните органи, особено дълги и скрити, например, хламидия, трихомониаза, микоплазмоза, уреаплазмоза, цитомегаловирусна инфекция, херпес.
- Неспецифични възпаления на простатата, уретрата и семенните мехурчета.
- Липса на епидидима, в която сперматозоидите „узряват“, и каналът, през който те излизат от тестиса.
- Запушване на семепровода или отстраняването им.
И накрая, има възможности, когато не е възможно да се установят причините за спада на репродуктивната функция, т.нар. Идиопатично безплодие. Във всеки случай, за да получите окончателна диагноза, е необходимо да се консултирате с уролог и да се подложи на пълен преглед.
Захарен диабет тип I, а именно, той се среща при деца, се отнася до заболявания, податливостта към които се наследява. Това е предразположението, но не самата болест, която се развива само когато провокира външни и вътрешни фактори, които действат върху тялото на бебето. Досега не са идентифицирани всички причини за диабета, но се смята, че инфекцията или стресът е най-често началната точка на заболяването.

Изглежда, че ако родителите или други роднини на детето са наясно с причините и признаците на диабет, тъй като самите те страдат, не трябва да има затруднения в ранното откриване на заболяването при детето. Въпреки това, за съжаление, най-често заболяването се намира в
късен етап, когато бебето вече е в тежко състояние и се нуждае от интензивни медицински дейности. Без значение колко тъжно е да се каже, на този етап, диабетът е много труден за лечение и причинява тежки усложнения. Ситуацията се усложнява от факта, че диабетът има много кратък скрит период, което означава, че няма време за дълги медитации.
Всеки възрастен член на семейството трябва ясно да знае първите признаци на диабет при децата и да може да оцени трезво здравните показатели, които са най-важни от гледна точка на ранната диагностика на заболяването.
Какви признаци на диабет трябва да забележат родителите?
- Промяна в апетита:
- появата на неестествено желание за детето;
- често е жажда, т.е. детето поради силното чувство на глад трудно може да издържи традиционните 3-4 часови паузи между храненията;
- слабост и сънливост след 1,5-2 часа след хранене.
Разбира се, повечето деца обичат сладкиши и мнозина са склонни да спят след хранене, но ако има генетична предразположеност към диабет тип I, тези навици могат да бъдат първите признаци на заболяването, а изолираното им съществуване може да означава, че болестта не е отишла далеч, така ще бъде по-лесно да се лекува. - Детето губи тегло, въпреки нормалния и дори повишен апетит.
Не е необходимо да се отписва загуба на тегло от факта, че детето се разраства бързо, по-добре е да го изследва и да се увери, че промяната в телесното тегло се дължи на повишена физическа активност и нарастващите нужди на детето, а не на факта, че тялото му се бори за борба с болестта. Въпреки че кръвта съдържа излишък от глюкоза, клетките на тялото на детето изпитват тежък глад, тъй като панкреасът със захарен диабет едва ли синтезира инсулина, необходим за абсорбирането на глюкозата от тъканите. - Детето бързо се уморява, става бавно, сънливо.
При липса на треска, кашлица и други признаци на простуда - тези симптоми трябва да бъдат предупредени от гледна точка на диабета. - Детето започва да пие повече и да уринира по-често, въпреки че този симптом е признат от диабетолозите късно.
Като сол, захарта привлича течност към себе си, тялото, опитвайки се да „разрежда” захарта, изисква вода, сигнализирайки го с жажда. Рано или късно, при захарен диабет, бъбреците спират да задържат глюкозата в тялото, тя започва да се екскретира с урината, което от своя страна води до увеличаване на диурезата. Децата с диабет започват да стават през нощта в тоалетната, а понякога и мокрят леглото. - Урината петна по гърне, в тоалетната, на памперси стане лепкава.
Това е чисто физическо явление, разтвор на захар, по очевидни причини, след изпаряването на водата оставя зад себе си лепкави петна. Внимателната майка винаги ще забелязва този симптом.
гадене, повръщане, коремна болка, суха и сърбяща кожа, признаци на невродермит, персистираща фурункулоза, пиодермия, замъглено виждане са късни симптоми и ефекти на диабета, знак, че болестта вече е набрала сила и ще бъде изключително трудно да се спре развитието му. Но ако родителите се обърнат към ендокринолог, когато се появят първите сигнали за диабет, болестта може да бъде открита в ранните етапи, дори преди да се наруши работата на панкреаса и нивото на захар в кръвта да се повиши. Времето няма да се пропусне и лекарите ще могат да спасят детските сили за борба с болестта и за пълноценен живот.
Внимание! Всички възрастни трябва да са наясно с наличието на диабет при дете: преподаватели в контакт с него, учители, съседи, приятели. Първо, това ще позволи да се избегнат грешки в храненето на бебето, второ, в случай на внезапно влошаване на здравословното състояние, да се предаде в специализирано ендокринологично отделение, а не в болница за инфекциозни болести или хирургия.
Сред наследствените заболявания, развиващи се в резултат на мутации, традиционно има три подгрупи:
- моногенни наследствени заболявания
- полигенни наследствени заболявания
- хромозомни аберации
От наследствени заболявания е необходимо да се разграничат вродени заболявания, причинени от вътрематочно увреждане, причинено например от инфекция (сифилис или токсоплазмоза) или от влиянието на други увреждащи фактори върху плода по време на бременността.
Много генетично обусловени заболявания се проявяват не веднага след раждането, а след известно време, понякога много дълго.
Моногенни наследствени заболявания
Моногенните болести се наследяват в съответствие със законите на класическата генетика на Мендел. Съответно за тях генеалогичните изследвания позволяват да се разкрие един от трите вида наследяване: автозомно доминантно, автозомно рецесивно и сексуално наследяване.
Това е най-голямата група наследствени заболявания. В момента са описани повече от 4000 варианта на моногенни наследствени заболявания, преобладаващата част от които са доста редки (например, честотата на сърповидно-клетъчна анемия е 1/6000).
Широка гама от моногенни заболявания образуват наследствени метаболитни нарушения, появата на които е свързана с генна мутация, контролирайки синтеза на ензими и причиняващи дефицит или дефект в структурно-ферментопатията.
Полигенни наследствени заболявания
Трудно е да се наследят полигенни заболявания. За тях въпросът за наследството не може да бъде решен въз основа на законите на Мендел. По-рано такива наследствени заболявания се характеризираха като болести с наследствена предразположеност. Тези заболявания включват заболявания като рак, диабет, шизофрения, епилепсия, коронарна болест на сърцето, хипертония и много други.
Хромозомни аберации
Хромозомните заболявания са причинени от грубо нарушение на наследствения апарат - промяна в броя и структурата на хромозомите. Типична причина, по-специално, е алкохолната интоксикация на родителите при зачеването („пияни деца“). Те включват синдрома на Даун, Клейнфелтер, Шерешевски - Търнър, Едуардс, "котешки плач" и други.
Диагностика и лечение на наследствени заболявания
Напоследък се твърди, че относително високата честота на наследствените заболявания се дължи на определени предимства на „мутанти“ по отношение на факторите на естествения подбор или „чувствителността към болести“.
Терапията на наследствени заболявания включва симптоматично лечение и генна терапия.
Симптоматично лечение
Наследствените заболявания се характеризират с различни симптоматични прояви и тяхното лечение е в много отношения симптоматично. Индивидуалните метаболитни нарушения правилно назначават специални диети, насочени към намаляване на токсичните вещества в организма, чието натрупване се дължи на мутации в определени гени. Например, когато фенилкетонурия предписва диета без беланин.
За да се облекчат симптомите на наследствени заболявания, свързани с дефект в определен протеин, интравенозно се налага функционалната му форма, която не предизвиква имунен отговор. Такава заместителна терапия се използва при лечение на хемофилия, тежка комбинирана имунна недостатъчност и др. Понякога, за да се компенсират определени загубени функции, се трансплантират костен мозък и други органи. Съществуващата терапия, за съжаление, в повечето случаи не е много ефективна.
Генна терапия
Принципно нов метод, ефективен и насочен към унищожаване на генетичната причина за наследствено заболяване, е генна терапия. Същността на метода на генната терапия е въвеждането на нормални гени в дефектни клетки.
Концепцията за генната терапия се състои в това, че най-радикалният начин за справяне с различни видове заболявания, причинени от промени в генетичното съдържание на клетките, трябва да бъде лечение, насочено пряко към коригиране или премахване на генетичната причина за самата болест, а не неговите последствия.
Поради факта, че генната терапия е нова посока на медицинската генетика и болестите, които се опитват да бъдат третирани по този начин, са много разнообразни, са създадени много оригинални подходи към този проблем. В момента изследванията на генната терапия са насочени основно към корекция на генетичните дефекти на соматичните, а не на зародишните клетки, което е свързано с чисто технически проблеми, както и от съображения за сигурност.
Според статията "Наследствени болести"
 Какво представляват наследствените човешки заболявания
Какво представляват наследствените човешки заболявания Киплинг - Приказки - текст с буквата Ё
Киплинг - Приказки - текст с буквата Ё Поздравления за Свети Валентин на английски език
Поздравления за Свети Валентин на английски език