Нарушаване на хемостаза. Повишена способност на кръвта да образува съсиреци в съдовете. Саон-антагонисти
Системата за хемостаза е забележително еволюционно постижение, което постоянно поддържа баланс между два различни насочени процеса: най-бързото образуване на съсирек (тромб), за да се предотврати загуба на кръв в отговор на увреждане на съда и запазване на течното състояние на кръвта в кръвообращението. Решението на тази трудна задача се осигурява от комплексни взаимодействия на съдовия ендотелиум, плазмената коагулационна каскада, антикоагулантните механизми, фибринолитичната система, тромбоцитите и левкоцитите. Стойността на реологичните характеристики на движението на кръвта през съдове с различни диаметри е голяма, особено вискозитета в микроциркулационната система.
В медицината често се развиват критични състояния на хемостазни нарушения, степента на тяхната тежест зависи от дерегулиращия ефект върху хомеостазата на увреждащите фактори - травма, инфекция, хирургия, лекарства, както и компенсаторните възможности на сърдечно-съдовата и дихателната системи. Отличителна характеристика на съвременната медицинска практика е дешифрирането на водещата роля на хемостатичните нарушения, по-специално синдромът на дисеминираната интраваскуларна коагулация (DIC) в патогенезата на най-критичните състояния, изискващи интензивна терапия, и появата на ефективни целеви средства за нейната корекция в арсенала на лекаря.
Клинично, хемостазните нарушения се проявяват по-често с кървене, рядко при тромбоза, но често се наблюдава едновременно проявление на патологично кървене и микротромбоза.
Физиология на нормалната хемостаза
Увреждане на стената на кръвоносните съдове причинява незабавна вазоконстрикция на самия наранен съд, както и съседните капиляри и артериола, което води до първоначално забавяне на притока на кръв в зоната на увреждане. Освен това, взаимодействието на няколко функционални компонента води до образуването на първичен тромбоцитен щекер (съсирек), който бързо се стабилизира чрез фибринови нишки. При нормални условия този прокоагулантен процес е ограничен по време и място, контролиран е от същите функционални компоненти. Те включват тромбоцити, плазмена коагулационна каскада, естествени антикоагуланти, фибринолизна система и ендотелни клетки.
Когато настъпи увреждане на ендотела, те влизат в контакт с субендотелиалния слой на колаген, настъпва адхезия (адхезия) на колагена, образуват се тромбоцитни гранули за освобождаване на серотонин, лизозомни ензими, фибриноген, тромбоцит и светлинна тъкан, се освобождава колаген. Активирани тромбоцитни агрегати за образуване на първична тромбоцитна кухина (първична хемостаза). Количествените и качествени патологични промени в тромбоцитите се проявяват чрез кървене и кръвоизлив. Времето на кървене е оптималният скринингов тест за оценка на функционалната жизнеспособност на тромбоцитите.
Коагулационната каскада е отговорна за образуването на стабилен фибринов тромб. Факторите, които я съставят, са изброени в Таблица. 2-10.
Таблица 2-10. Коагулационни фактори Фактор 1 Фибриногенен фактор II Протромбинов фактор III фактор
Повечето фактори на кръвосъсирването се синтезират в черния дроб. Фактор VIII, в допълнение към черния дроб, частично синтезира мегакариоцити и ендотелни клетки. Фактори II, VII, IX и X витамин К са зависими, т.е. Витамин К е необходим за техния синтез чрез хепатоцити, а на фиг. 2-8.
Началото на коагулационната каскада е взаимодействието на фактор VII с тъканния фактор (III) в зоната на увреждане (външен път на коагулация). Полученият комплекс от активиран фактор VII с тъканен фактор активира фактори IX и X, което води до образуването на тромбин. Тромбинът преобразува фибриноген в фибрин, активиращ фактор XIII и тромбоцити. Действието на тромбина е под контрола на естествен антикоагулант - активиран протеин С. Алтернатива на външния път на коагулация е вътрешен, започващ с активиране на фактор XII от колаген, дължащ се на контакт на кръв с извънземна повърхност. XHa фактор активира фактор X1a, след това външните и вътрешните пътища на коагулация са идентични.
Вътрешен път Външен път
(фактор на контакт) (увреждане на тъканта)
RSH
PXA
Rua
RHSha
RHSH
Фиг. 2-8. Схема на коагулационна каскада.
Скрининговите тестове - протромбиново време, активирано парциално тромбопластиново време (АРТТ) и тромбиново време - позволяват да се прецени състоянието на коагулационните хемостазни единици. Вродени или придобити дефекти на коагулационната каскада причиняват кръвоизливи в мозъка, ставите, меките тъкани и мускулите и стомашно-чревно кървене.
Естествените антикоагуланти са представени от два основни фактора - антитромбин III и витамин К зависими протеини С и 5. Антитромбин III инхибира тромбина и фактора Ха, участва в инактивирането на фактори 1Ха, Х1а, XIa. Чрез взаимодействие с хепарин, антитромбин III значително подобрява антикоагулантния му ефект. Активираният протеин С във връзка с протеин 5 има антикоагулантно действие върху ензимите, които инхибират фактори UA и USH (важни кофактори на прокоагулантния процес). В допълнение, протеин С усилва фибринолизата чрез инхибиране на тъканния плазминогенен активатор, улеснявайки прехвърлянето на плазминоген в плазмин. Протеин С играе важна роля в патогенезата.
DIC синдром. Недостигът на антитромбин III, протеин С и протеин 5 (по-често придобити от вродената) е съпроводен с висока честота на тромботични усложнения.
Фибринолизата (подобно на коагулацията) е нормален отговор на увреждане на съдовата стена. Тъканният активатор на плазминоген (1-РА), образуван от ендотелни клетки в отговор на тяхното увреждане или стимулиране с тромбин, превръща плазминогена в плазмин. Плазминът разрушава фибрина и фибриногена, образувайки различни продукти на разграждане на фибрина. В същото време, инхибитор на плазминогенен активатор (PA1-1, образуван от хепатоцити и ендотелни клетки и PA1-2, образувани в плацентата и макрофагите) циркулира в неактивна форма в плазмата. Тяхното физиологично значение е в контролирането на фибринолизата и предотвратяване на прехода й към патологичния стадий. Терапевтичните инхибитори на фибринолизата са транексамова киселина и аминокапронова киселина. Обратно, стрептокиназата е фибринолитично лекарство, което увеличава образуването на плазмин.
Ендотелните клетки при нормални условия са отговорни за антитромботичното взаимодействие между кръвта и тъканите, поддържайки течното състояние на кръвта. Те произвеждат антикоагуланти като гликозаминогликани, хепарин сулфати, тромбомодулин (активират антитромбин III и протеин С), азотен оксид и простагландин (инхибират агрегацията на тромбоцитите и стимулират вазодилатацията), тъканен плазминогенен активатор, който инициира фибринолиза. Но ендотелните клетки, в отговор на стимулиране от бактериални ендотоксини, са способни да произвеждат фактор на Willebrand и тъканни фактори на тяхната повърхност, които задействат коагулационната каскада. Тези свойства трябва да бъдат взети под внимание при лечение на нарушения на хемостазата.
Лабораторен скрининг за хемостатични нарушения
Тестовете за оценка на първичната хемостаза, извършена спешно при условия на интензивна терапия, са определянето на времето на кървене и броя на тромбоцитите. Агрегирането и адхезията на тромбоцитите, съдържанието на фактора на von Willebrand се изследват в специализирани коагулологични лаборатории.
Процесът на коагулационна хемостаза може да бъде оценен чрез тестове като APTT, протромбиново време и тромбиново време, които дават пълна оценка на някои фактори на кръвосъсирването. Отделно определяне на коагулационните фактори е възможно само в специализирани лаборатории.
Скринингът на антикоагулантната система може да се извърши чрез определяне на концентрацията на антитромбин III; Други тестове за инхибиране на коагулацията отнемат много време, те се извършват в специализирана лаборатория.
Състоянието на фибринолитичната система може да се прецени чрез изследване на количеството на продуктите от разпадането на фибриногена, фибриновите мономери, В-димерите, както и използването на специфични изследвания на съдържанието на активатори на фибринолиза и инхибитори в плазмата.
Нормалните стойности на параметрите на коагулацията са дадени в таблица. 2-11.
Таблица 2-11. Параметри на коагулацията на кръвта Норма на теста APTT 27.4-40.3 s Протромбиново време 12.3-16.1 s Контрол на тромбиновото време ± 3 s Фибриноген 1.7-3.1 g / l Р-димери Таблица. 2-11 Коагулационни фактори II, V, VII, VIII, IX, X, XI, XII 50-150% Антитромбин III 80-120% Активен протеин С 73-121% Протеин 3: общо 55-125% свободен 21-53% Willebrand фактор (антиген) 50-150%
Времето на кървене съответства на времето, необходимо за образуването на тромбоцитния щекер. Той може да бъде удължен с тромбоцитопения, тромбоцитна дисфункция и болест на фон Вилебранд, но никога не се удължава с нарушения на коагулацията (например, хемофилия).
Броят на тромбоцитите трябва да бъде включен в задължителния лабораторен скрининг при пациенти в интензивни отделения. Трябва да се помни, че броят на тромбоцитите може да бъде намален в първите дни на менструацията.
APTT отразява общото съдържание на всички фактори на вътрешния път на коагулация - от активирането на фактор XII до образуването на разтворим фибрин, с изключение на фактори VII и XIII. Удължаването на АРТТ отразява повишена склонност към кървене и може да показва дефицит в един или повече фактори на вътрешната връзка на хемостазата (например, VIII при хемофилия или болест на Willebrand). AHTV удължава терапията с натриев хепарин или варфарин. Нарушенията на първичната хемостаза не отразяват APTT.
Протромбиновото време характеризира образуването на съсирек. Той определя сумата на коагулационните фактори I, II, V, VII и X, съставляващи външния път, и удължава с дефицит на една или повече от тях. Този индикатор често се използва за контрол на терапията с непреки антикоагуланти (варфарин) - антагонисти на витамин К, които съответно не действат върху фибриноген и фактор V. Изследването на протромбиновия комплекс, който определя само сумата от фактори II, VII и X (за синтеза на тези фактори се нуждаят от витамин к). За да се сравнят резултатите от изследванията на различни лаборатории, използващи различни тестови системи, показателите за протромбиново време и протромбиновия комплекс се преизчисляват в международно нормализирано съотношение (INR). При здрави индивиди INR е около 1. При лечение с варфарин, дефицит на витамин К, чернодробна недостатъчност или изолиран дефицит на един от факторите (обикновено VII), стойностите на INR трябва да бъдат между 2 и 3.
Тромбиновото време е тест за крайния стадий на тромбоза. Удължаването му може да показва фибриногенеза под 1 g / l или дисфибриногенемия. Терапията с хепарин натрий също удължава тромбиновото време.
Ниското съдържание на фибриноген може да се дължи на намалено производство или увеличен прием. Високото съдържание на фибриноген е индикатор за остро възпалително състояние, особено на черния дроб, където се синтезира. Увеличаването на концентрацията на продуктите на либриногенен лизис (продукти на разграждане на фибрин, В-димер), докато намалява неговото количество, показва развитието на DIC.
Недостигът на антитромбин III (най-силният природен протеолитичен плазмен антикоагулант) често е проява на остра масова загуба на кръв, неадекватно попълнена [без преливане на прясно замразена плазма (FFP)] или DIC, придружаваща сепсис. Много по-рядко, намаляването на количеството на антитромбин III се проявява като проявление на наследствено заболяване с автозомно доминантна природа. Използването на скринингови тестове за диференциалната диагноза на придобитите хеморагични синдроми или заболявания, чиято основна клинична проява е патологията на хемостазната система, е показана в Таблица. 2-12.
Таблица 2-12. Изследване на хемостаза при някои заболявания и синдроми Тестова хемофилия
Von Willebrand Пикантен
двигател с вътрешно горене
синдром на болестта
хепарин
натриев варфарин тромбоцити Норма Норма Намалена Норма Норма Норма Норма на фибриногена Намалена Намалена Норма Норма Норма Протромбин
време Норма Норма Удължена Удължена Норма Удължена АРТТ Удължена Норма или удължена Норма или удължена Норма или удължена Удължена Удължена Тромбиново време Норма Норма Удължена Норма или удължена Норма
Въпреки това, изброените класически тестове за мониторинг на хемостаза, за съжаление, не дават интегрална, цялостна картина на неговото състояние, отразяваща взаимодействието на множество фактори. В интензивното лечение често е необходимо да се знае за състоянието на кръвосъсирващата система при пациент по време на проучването, както с цел да се изберат средства за медикаментозна или трансфузионна корекция, така и с цел да се оцени коректността на проведената терапия за коригиране на хемостазните нарушения. Методът на тромбоеластографията позволява бързо и надеждно получаване на данни, характеризиращи както общото състояние на хемостазата, така и състоянието на отделните му връзки с патологичните им промени.
Методът на тромбоеластографията се основава на измерване на физичните параметри на съсирек (тромб) по време на образуването му. От момента на започване на коагулацията и до неговото прекратяване и последващия лизис с помощта на електромеханичен преобразувател се регистрира промяна в плътността на съсирека, която в съвременните модели на тромбоеластографа се предава на компютърен дисплей. Различават се следните основни параметри на тромбоеластографията (фиг. 2-9):
К (мин) е времето от началото на коагулацията до образуването на първите фибринови влакна;
K (min) - времето на промяната в коагулационната амплитуда, нейното увеличаване или намаляване;
К + К - корелира с времето на коагулация, равно на 6-8 минути в норма;
ъгъл а - отразява скоростта на кръвосъсирването, процеса на полимеризация на фибрина;
MA (mm) - максималната амплитуда на кривата, характеризираща силата и твърдостта на образувания съсирек, които зависят главно от функцията и броя на тромбоцитите и в по-малка степен от концентрацията на фибрин в кръвта;
ЕДЬ, БУЗО (%) - оценка на степента на фибринолиза.
Както може да се види от фиг. 2-9, с хиперкоагулационен синдром, времевите индекси на KIC се скъсяват, ъгълът a и MA се увеличават. Напротив, със статуса на хипокоагулацията, C & Cs нараства, ъгълът a и MA намаляват. В допълнение към общите характеристики на състоянието на хемостаза, според данните на тромбоеластографията, е възможно да се преценят и нарушенията на отделните му връзки. Така, в случай на предозиране на натриевия хепарин, всички параметри на тромбоеластограма ще бъдат удължени и намалени - K, K, a, MA; за тромбоцитопения, К ще остане в нормалните граници, но К ще бъде удължен с намалена МА; в случай на засилено фибринолиза, # е нормално, но МА е рязко и дългосрочно редуцирано до EPI и LUZO; тромбоцитопатия и тромбоцитна дисфункция
начало
Фиг. 2-9. Варианти на тромбоеластограма: А - норма; В - хиперкоагулация; Б - хипокоагулация.
К е удължен, ъгълът а и МА са намалени. Възможността за повторна тромбоеластография, нейното графично записване, минималното време за изследване с много малко количество кръв, необходими за нейното прилагане, правят тромбоеластографа един от съществените атрибути на лабораторното оборудване на експресната лаборатория на интензивните отделения.
ИНТЕНЗИВНА ТЕРАПИЯ НА ОТДЕЛНИТЕ ФОРМИ НА РАЗСТОЯНИЕ НА СИСТЕМАТА НА ХЕМОСТАЗА
В критична медицина, пълно проучване на коагулацията често не може да бъде проведено поради тежестта на състоянието на пациента, липсата на време и спешността на хирургичната интервенция. Трябва да се отбележи, че в преобладаващата част от случаите може да се изключи съмнението за наличие на кървене, дължащо се на системна хемостаза, ако пациентът има хематокрит\u003e 30%, брой на тромбоцитите\u003e 100x109 / l, концентрация на фибриноген\u003e 1,5 g / l и едновременно APTT и протромбиновото време е не повече от 3-5 s по-високо от горните граници на нормалните стойности. С такива показатели е възможно безопасно да се извършват инвазивни процедури (катетеризация на централната вена или пункция на артерията). В същото време, кървенето при тези скорости може да показва дефект в хирургичната локална хемостаза (обикновено след операция) или развитието на остър DIC.
Най-често срещаните системни нарушения на хемостазата, които изискват интензивно лечение, са дадени в Таблица. 2-13.
Таблица 2-13. Нарушения на хемостаза при критични състояния Диагноза Патология на хемостаза Чернодробно заболяване Намаляване на всички фактори на кръвосъсирването, с изключение на фактора VII и von Willebrand Намалена концентрация на протеини C и 5 Намален брой тромбоцити и тяхната дисфункция DIC синдром Dysfibrinogenemia opn Разстройство на тромбоцитите Намаляване на антитромбозата, за 15 минути използване на AIC Тромбоцитна дисфункция Намаляване на броя на тромбоцитите Намаляване на концентрацията на фибриноген участници II, V, VII, X, XI DIC-синдром TBI, катастрофа-синдром Увеличаване на концентрацията на продуктите от разграждане на фибрин DIC-синдром Масивни трансфузии Намаляване на съдържанието на фактори V и VII DIC-синдром Варфарин Намаляване на съдържанието на фактори II, VII, IX, X Намаляване на концентрацията Протеин С и 5 Натриев хепарин Тромбоцитопения Намаляване на съдържанието на фактор X тромболитичен
терапия Повишаване на концентрацията на фибрин и продукти на разграждане на плазмин Намаляване на съдържанието на фибриноген DIC синдром Намаляване на съдържанието на фибриноген Намаляване на броя на тромбоцитите Намаляване на съдържанието на протеин С и 5 Намаляване на концентрацията на антитромбин III Намаляване на съдържанието на фактори V, VIII, IX, XI Увеличаване на концентрацията на фибринови продукти \\ t
Минималното достатъчно съдържание на фактори на плазмената коагулация в кръвния поток, което е необходимо, за да се осигури хемостаза по време на хирургични интервенции, е илюстрирано в Таблица. 2-14.
Минималният брой тромбоцити е 80-100x109 / l.
Таблица 2-14. Минимално съдържание на коагулационни фактори, необходими за хирургичния хемостаза Фактор Хемостатично ниво,% от нормалния фибриноген 50-100 протромбин 40-50 Фактор V 10-30 Фактор VII 10-20 Фактор VIII 30-70 Willebrand фактор 20-50
В края на таблицата. 2-14 Фактор IX 20-60 Фактор Х 10-20 Фактор XI 20-80 Фактор XIII 10
Нарушения на първичната хемостаза
Тромбоцитопенията (виж глава 9), болестта на фон Вилебранд и дисфункцията на тромбоцитите се отнасят до хеморагична диатеза, причинена от увреждане на първичната хемостаза. Типични симптоми - хеморагичен обрив по кожата или лигавиците, поява на синини, петехии с минимално въздействие. Необходима е интензивна терапия за местно кървене, особено често с назална и менорагия.
Най-често се регистрира тромбоцитопения, дължаща се на увреждане на костния мозък (апластична анемия, хемобластоза, ракови метастази). Рядко в практиката на интензивен лекар има пациенти, чиято хеморагична тромбоцитопенична диатеза се дължи на вродени причини (синдром на Wiskott-Aldrich, май-хеглин аномалия) или радиационно увреждане, железен дефицит, витамин В12, хроничен алкохолизъм.
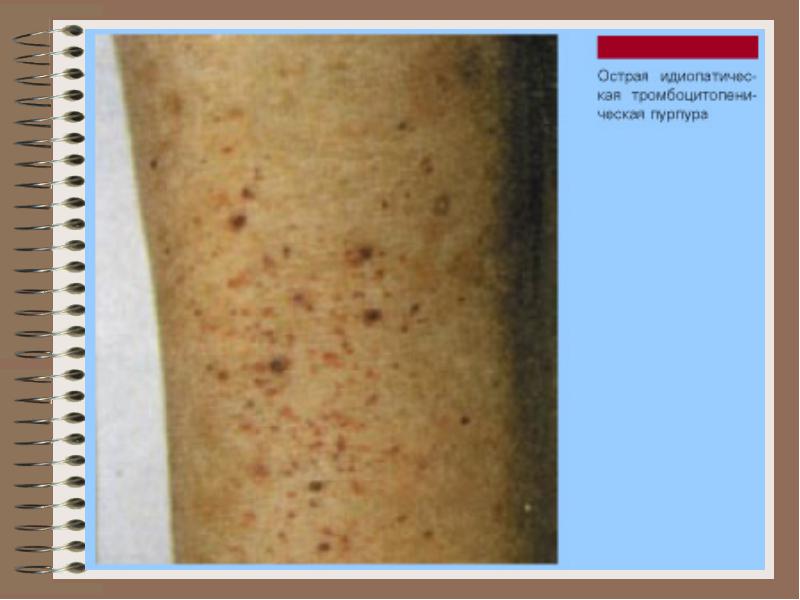
Идиопатичната тромбоцитопенична пурпура е най-честата форма на автоимунна тромбоцитопения, която се характеризира със скъсяване на живота на тромбоцитите поради повишената им консумация. Функцията на тромбоцитите не страда. Когато се появи кървене или за да се предотвратят тях, се предписват глюкокортикоиди. Ефективно използване на високи дози интравенозен имуноглобулин. Често, особено когато се появяват усложнения от глюкокортикоидната терапия (синдром на Иценко-Кушинг, хипергликемия) или рецидивиращо кървене, те прибягват до спленектомия. В деня на операцията и в непосредствения следоперативен период, продължава терапията с глюкокортикоиди, нейното отменяне трябва да бъде постепенно (намаляване на дозата от 2.5-5 mg преднизолон на ден). Тромбоцитните трансфузии не се считат за средство за избор, освен в изключително редки случаи (неефективността на консервативната терапия и необходимостта от предотвратяване на повишено кървене по време на операцията).
Имунният конфликт служи и като основа за хепарин-индуцирана тромбоцитопения. Тялото на пациента, в отговор на въвеждането на натриев хепарин, започва да образува антитела 1cC, които се свързват с тромбоцитните рецепторни компютри, като допринасят за съкращаването на живота им. Честотата на възникване на такъв конфликт -
5% от индивидите, получаващи хепарин натрий. Тромбоцитопенията се развива бавно, локалното кървене е рядко, тромбоцитни трансфузии не са необходими. С премахването на хепарин се възстановява броят на тромбоцитите в рамките на 2-5 дни.
Много лекарства, използвани в интензивното лечение, могат да причинят нарушена функция на тромбоцитите, но обикновено са клинично незначителни. Малцина могат да причинят кървене.
Ацетилсалициловата киселина блокира тромбоцитната агрегация, инхибирането продължава през целия живот на тромбоцитите в кръвообращението (7-10 дни), затова дори една доза ацетилсалицилова киселина може да блокира основната функция на първичната хемостаза за доста дълго време. Ако е необходимо да се извършат операции в такава ситуация, се показва предписването на десмопресин, което временно (за три часа) елиминира удължаването на времето на кървене. Повторното назначаване на десмопресин удължава действието му.
НСПВС блокират синтеза на тромбоксан А2 - важен медиатор на тромбоцитната агрегация. Тяхното действие може също да бъде неутрализирано чрез предписване на десмопресин.
Разтворите на декстран намаляват плазмената концентрация на фактор VIII, фактора на von Willebrand, инхибират тромбоцитната функция, удължават времето на кървене. Въпреки доброто си обемно-заместващо действие, трансфузия на декстрани при остра масова загуба на кръв или при първоначално интензивна хемостаза (хемофилия, заболявания на кръвоносната система, чернодробни заболявания и др.) Трябва да бъде сведена до минимум.
Клопидогрел, тиклопидин, както и съвременните тромбоцитни рецепторни инхибитори, СРПЬ / Ша (abciximab), използвани при пациенти с коронарна болест на сърцето (CHD), които са претърпели инсулт или инфаркт на миокарда, причиняват дисфункция на тромбоцитите и увеличават риска от хеморагични усложнения. В почти всички случаи, прилагането на десмопресин помага в тези случаи, с изключение на ситуацията с употребата на инхибитори СРНЬ / Ша. В последния случай може да е необходимо преливане на тромбоцити.
Заболяването на Von Willebrand е хеморагична диатеза, наследена от автозомно доминантния тип, причинена от дефект или липса на фактор на von Willebrand в плазмата. Факторът на von Willebrand има две основни функции. Той е необходим за образуването на тромбоцитна тапа и предпазва фактор VIII от разцепване в плазмата. Съществуват три основни вида болест на von Willebrand (Таблица 2-15).
Таблица 2-15. Болест на Willebrand: видове и терапия Факторът Willebrand Терапия Количествен дефект Десмопресин в повечето случаи Качествен дефект Десмопресин в леки случаи; концентрати на фактор VІІІ, съдържащи фактор на Willebrand; криопреципитат Общо отсъствие на концентрати на фактор VIII, съдържащ фактор на Willebrand, криопреципитат
Епидемиологичните проучвания показват, че честотата на болестта на Willebrand не надвишава 1% в популацията, но обикновено се диагностицира по-рядко. Болестта преобладава сред жените с менорагия. При всички пациенти с кървене трябва да се определи количеството на фактора на von Willebrand в кръвта.
Първият вид заболяване е най-често срещан, той съставлява 70-80% от всички лица, страдащи от болестта на фон Вилебранд. При тази форма на заболяването броят на нормално функциониращия фактор на Willebrand намалява. Тенденцията към кървене обикновено се изразява умерено, но има и тежки прояви.
Вторият тип се характеризира с качествен дефект на фактора Вилебранд, дължащ се на мутация в гена на този фактор.
Третият тип се характеризира с липса на фактор на von Willebrand и намаляване на концентрацията на фактор VIII в плазмата.Диагностиката обикновено се основава на лабораторни изследвания, главно на определяне на концентрацията и функцията на фактора на Willebrand и на активността на фактор VIII.
Терапията е насочена към нормализиране на три параметъра - съдържанието на факторите на von Willebrand и VIII, времето на кървене. Това се постига чрез прилагане на десмопресин, който стимулира ендогенна хемостаза, и чрез прилагане на концентрат на фактор VIII, съдържащ фактор на von Willebrand. Десмопресин е ефективен в първия, по-рядко при втория тип болест на фон Вилебранд и е неефективен в третия тип. Когато се прилага интравенозно, ефектът е практически фулминантен, с необходимост от продължаване на лечението, повторни дози се прилагат след 8-12 часа, а при втория и третия тип болест на фон Вилебранд и при отсъствие на ефекта на прилагане на десмопресин, в първия тип, се провежда терапия с фактор VIII концентрати, съдържащи фактор Willebrand. Не се използват чисти концентрати на фактор VIII, тъй като фактор VIII има много кратък полуживот в отсъствието на фактора на von Willebrand. Всички концентрати на фактор VІІІ, съдържащи фактор на Вилебранд, се получават от плазмата, следователно, концентрати, които са претърпели вирусна инактивация, трябва да се използват, за да се избегне инфекция чрез трансфузия чрез вирусни инфекции. Именно вирусната несигурност на криопреципитата ограничава употребата му при лечението на болестта на фон Вилебранд. Понякога, особено при изразено кървене на лигавиците, те прибягват до назначаването на инхибитор на фибринолизата - прилагане на транексамова киселина.
Антикоагулантите, дезагрегираните, декстранните трансфузии са противопоказани за хора с болест на von Willebrand. Те не трябва да получават интрамускулни инжекции, тъй като рискът от интрамускулни хематоми е висок.
Нарушения на коагулационната каскада
При заболявания, причинени от патологията на коагулационната каскада, образуването на фибрин в крайна сметка е нарушено. При вродената природа на заболяването, в плазмата няма един единствен фактор на кръвосъсирването, докато при придобитите заболявания има няколко недостатъка (например вторична хемостаза, дължаща се на чернодробно заболяване, дефицит на витамин К, използване на антикоагуланти).
Хемофилия AIV
Хемофилията е вродена болест на системата на кръвосъсирване, наследена от рецесивен тип, свързан с пола и причинена от дефицит на фактор VIII (хемофилия А) или фактор IX (хемофилия В). Клинично тези две форми на заболяването са идентични, те се диференцират само при изследване на концентрацията на всеки фактор в кръвта.
Честотата на хемофилията А е една на 5000 новородени момчета, хемофилия В - един на 30 000 новородени момчета. Съответно, 80% от пациентите с хемофилия имат хемофилия А, 20% имат хемофилия Б.
Гените на фактори VIII и IX са свързани с женската Х хромозома, така че хемофилията засяга само мъжете, а жените стават носители. При женските превозвачи рискът от момче с хемофилия е 25%, а на момиче-носителка също 25%. При мъжете с хемофилия дъщерята винаги ще бъде носител, а синът винаги ще бъде здрав.
Клиничните прояви на хемофилия зависят от концентрацията на фактор VIII или IX в кръвта. Тяхната дейност се определя в международни единици (IU). Нормалната активност на фактори VIII или IX е около 1 IU в 1 ml плазма. Има три степени на тежест на хемофилията (табл. 2-16).
Таблица 2-16. Тежест на хемофилия Тежест Концентрация на фактор VIII, t / t Концентрация на фактор IX, IU / dl Лека хемофилия А 5-25 100 Умерена хемофилия А 1-4 100 Тежка хемофилия Обикновено е необходима интензивна терапия при пациенти с тежка хемофилия, при които спонтанни кръвоизливи и кръвоизливи могат да се появят във всички органи и тъкани, да са с повтарящ се характер. Кръвоизливи в ставите водят до развитие на хемофилна артропатия, в мускулите - те могат да причинят компресионен синдром, в меките тъкани - образуването на псевдотумор, който е капсулиран и бавно се увеличава в резултат на многократни кръвоизливи, изстисква околните тъкани. Вътречерепното кръвоизлив е една от най-честите причини за смърт при пациенти с хемофилия.
Лека форма на хемофилия често не се разпознава до зрялата възраст на момчетата, когато може да бъде открита за първи път, ако е необходимо да се извърши всякакъв вид операция. Тежката хемофилия обикновено се диагностицира, когато момчетата са на 1-2 години, когато започнат да ходят. Първите признаци могат да бъдат кръвоизливи в ставите, подкожни хематоми поради детски шеги или интрамускулни инжекции. В коагулограмата удължаването на APTTV се отбелязва при нормални стойности на протромбиновото и тромбиновото време, както и времето на кървене. Диагнозата се потвърждава от изследването на концентрацията на фактори VIII, IX и Willebrand.
За лечение се използват концентрати на фактори VIII или IX, получени от донорска плазма или чрез използване на генна технология. Съвременните концентрати на коагулационните фактори са безопасни за вируса и високо ефективни. При изчисляване на дозата на концентрата на фактора се приема, че кръвта на реципиента не трябва да се съхранява поне 50% в деня на операцията и по време на
14 дни от следоперативния период. Обикновено количеството IU на фактор VIII или IX, необходимо за еднократно прилагане, се изчислява, както следва:
обем на кръвта (ml) \u003d 7% от телесното тегло (kg); например 7% от 70 kg \u003d 4900 ml;
плазмен обем (ml) \u003d 60% обем на кръвта (ml); 60% от 4900 ml \u003d 2940 ml;
количеството ME фактор VIII \u003d 50% плазмен обем (ml); т.е. в нашия пример 1470 IU.
Многообразието на интравенозно приложение за концентрати на фактор VIII - всеки
h, за фактор IX - на всеки 12 часа
Приблизително 30% от пациентите с хемофилия А в процеса на лечение с фактор VIII се концентрират в кръвта и се появяват антитела, блокиращи прокоагулантната активност на лекарството и поради това се наричат \u200b\u200bинхибитори. При хемофилия В честотата на поява на инхибитора е по-малка. Титърът на инхибитора се измерва в единици Bethesda (BU). Една BU е равна на количеството инхибитор, инхибиращ 50% от една единица фактор VIII, въведен в тестовата система. Нисък титър на BU (5-10 BU) може да бъде преодолян чрез увеличаване на дозата на фактора, но високият титър на BU (над 10 BU) прави невъзможно получаването на хемостатичен ефект, тъй като цялото количество инжектирано лекарство ще бъде блокирано от антитела. За отстраняване на антитела се препоръчва плазмофереза \u200b\u200b(не замествайте отведената плазма на донорната FFP, тъй като тя съдържа фактор VIII), прилагането на глюкокортикоидни лекарства. Напоследък рекомбинантният активиран фактор VII и активираният концентрат на протромбиновия комплекс се използват широко в такива ситуации. При лечението на такива пациенти е задължителна консултацията с коагулолог.
ФАКТОР НА НЕДОСТАТЪЦИ VII
Коагулационният фактор VII, чрез свързване с тъканния фактор, се активира и става начален лост, от който започва процеса на коагулация. Много рядко се диагностицира генетичният вариант на вроден дефицит на фактор VII. Клинично се проявява с образуването на синини с леки наранявания, продължително несвързано назално и маточно кървене. При тежък дефицит на фактор VII (по-малко от 1%) може да се получи кървене, както при хемофилия, хемартроза, ретроперитонеални хематоми и интрацеребрални кръвоизливи. В същото време, APTTV остава в нормалните граници, но протромбиновото време и INR се увеличават. За крайната диагноза е необходимо да се изследва активността на фактор VII в кръвта.
Придобит дефицит на фактор VII се развива при чернодробни заболявания, лечение с варфарин, дефицит на витамин К. В същото време концентрацията на други фактори на кръвосъсирване, зависими от витамин К - II, IX, X, ще бъде понижена в кръвта.
Терапията е заместителна. Като източник на фактор VII се използва FFP трансфузия, предписва се концентрат на протромбиновия комплекс. Наскоро в арсенала на лекаря се появи рекомбинантен активиран фактор VII, който се е доказал при лечението на дефицит на фактор VII и при лечението на инхибиторни форми на хемофилия и тромбоцитопенично кървене. За да се постигне хемостаза, е необходимо да се увеличи концентрацията на фактор VII в кръвта на пациента до 15-20%. Препоръчителната доза рекомбинантно активиран фактор VII е 90-120 mg / kg интравенозно всеки път
3 часа, за да спре кървенето. Лабораторният мониторинг е необходим за мониторинг на ефикасността и корекцията на дозата, както и за елиминиране на възможния риск от тромбоемболични усложнения.
ДРУГИ СЪСТАВНИ ДЕФИЦЕНТИ НА ПЛАЗМЕНИТЕ ФАКТОРИ
Недостиг на фактор XI (хемофилия С) е вродена хеморагична диатеза, която е с подчертано етнически характер и се диагностицира главно сред евреи и арменци. Многократното кървене с различна локализация (назална, менорагия, следродилна, посттравматична) се развива, когато съдържанието на фактор XI е по-малко от 10%. APTTV е удължен, протромбиновото време е в нормални граници. Ако е невъзможно да се провери лабораторния дефицит на фактор XI в лабораторни условия с повишено кървене, е необходимо да се предпише преливане на донорната FFP. Целта на антифибринолитиков (tranexamic и аминокапронова киселина) е неефективна и е противопоказана при хематурия.
Афибриногенемията е много рядка хеморагична диатеза, проявяваща се с чести спонтанни тежки кръвоизливи. При хипофибриногенемия спонтанно кървене се появява само при концентрация на фибриноген, по-малка от 0,5 g / l. APTT и протромбиновото време са значително удължени. Диагнозата се потвърждава чрез лабораторно изследване на съдържанието на фибриноген. Терапия - преливане на FFP, криопреципитат или фибриногенен концентрат.
Дисфибриногенемията е хетерогенна група наследствени заболявания с фибриногенна дисфункция. Понякога тази дисфункция се проявява чрез резистентност на фибриновия съсирек до фибринолиза, по-често - свръхчувствителност към фибринолиза. В първия случай има тенденция към повишена тромбоза, а във втория - до кървене. Лечение след проверка на диагнозата (с участието на коагулолог) - FFP трансфузия, по-рядко - назначаване на ген фибрино.
Изолиран вроден протромбинов дефицит, фактори V, X,
XII, XIII в практиката на интензивното лечение е изключително рядко. Тези нарушения нямат специфична клинична картина, диагнозата им изисква не само скрининг проучвания на коагулограма, но и специфични анализи на концентрацията на специфични фактори на кръвосъсирването. При откриване на етиологичната причина за кървене, FFP се прелива.
АНТИФОСФОЛИПИДНИ СИНДРОМИ И АНТИКОАГУЛАНТИ НА ТРУДА
Кървене на лигавиците в комбинация с прояви на венозна или артериална тромбоза на фона на тромбоцитопения с едновременно откриване на антифосфолипидни антитела и / или лупус антикоагулант в кръвта и тежка форма - катастрофичен антифосфолипиден синдром. Този синдром се проявява със системен лупус еритематозус, колагеноза, рак, инфекции и бременност. Много случаи на катастрофичен антифосфолипиден синдром са идиопатични по природа. В основата на този синдром е образуването на автоантитела, насочени срещу фосфолипиди, по-специално кардиолипин. Последният образува комплекс с протеин (32-гликопротеин-1 (р2-СР1), който е част от протромбин. По тази причина, плазмата на пациенти с катастрофичен антифосфолипиден синдром има антикоагулантна активност, а съдържанието на лупусния антикоагулант може да се прецени по степента на тази активност. В случай на антифосфолипиден синдром, всички протромбин-зависими тестове ще бъдат удължени, докато въпреки удължаването на APTT и UIGO, времето на коагулация на t U1UO не се нарушава, което определя високия риск от тромбоза в случай на антифосфолипиден синтез. Dre.
Терапията за катастрофичен антифосфолипиден синдром е сложна. На фона на плазмафереза, предписан UFG, преднизон. С неефективността на провежданата импулсна терапия с метилпреднизолон. Отстранената плазма се попълва от донора FFP. Често при тотална тромбоза на мезентериалните съдове е необходимо да се прибегне до хирургична интервенция, което значително влошава прогнозата.
Цирроза на черния дроб
Цялостното нарушаване на хемостатичния баланс поради намаляване на синтеза на много фактори на кръвосъсирването и инхибитори при чернодробни заболявания е съпроводено с развитие на патологично кървене. Това намалява броя и функцията на тромбоцитите, увеличава фибринолизата, удължава протромбиновото време, увеличава INR. Често спленомегалията, наблюдавана при чернодробна цироза, влошава тромбоцитопенията. В случай на тежко кървене или хирургична интервенция, се предписва FFP трансфузия, приложение на рекомбинантен активиран фактор VII, протромбинов комплекс. Транексамовата киселина и десмопресин действат за кратко.
Разпространена интраваскуларна коагулация
DIC е пълно разрушаване на взаимодействието на всички системи на хемостатичен баланс, които определят неговото равновесие, което включва ендотелни клетки, тромбоцитна (първична) хемостаза, плазмена коагулационна система и фибринолиза. За ICE се характеризира с едновременно наличие на кървене и микротромбоза, което води до бързо развитие на органа и PON. Дешифрирането на явлението ICE (в което произведенията на MS Machabeli и ZS Barkagan играят водеща роля в нашата страна) обяснява много от привидно различни феномени, съпътстващи такива формално различни критични състояния като сепсис, масивна загуба на кръв, изгаряния и змийски ухапвания. усложнения при бременност и др.
В практиката на интензивното лечение ICD най-често се развива на фона на сепсис, масивна загуба на кръв (особено в акушерството), наранявания, рак и хематологични заболявания.
ЕТИОЛОГИЯ И МЕХАНИЗЪМ НА РАЗВИТИЕТО
Развитието на ICE може да се дължи на различни причини и условия, както е представено по-долу. По-често, ДВГ придобива характера на остър (мълния) курс, по-рядко се наблюдава хроничен ход.
Причини за DIC:
Шок (хиповолемия и хипоксия) от всякаква етиология.
Инфекция.
Сепсис.
Бактериален.
Вирусни.
Гъбична.
нараняване
Бърнс.
Синдром на катастрофата
-CHMT.
Дебел емболия.
Големи операции.
Усложнения на бременността и раждането
Тежка еклампсия.
Разкъсване на плацентата.
Фетална смърт на плода.
Емболия на амниотичната течност.
синдром NENR.
анафилаксия
обида
Остра интраваскуларна хемолиза
Съдова протеза
Ухапване от змия
неоплазми
Аденокарцином.
Хематологични злокачествени заболявания.
Чернодробно заболяване
Цироза.
Остър хепатит.
Патологичното активиране на хемостазната система може да бъде причинено от различни фактори. Активирането на ендотелни клетки от ендотоксин при инфекции, отравяне, ацидоза и хипоксемия е придружено от появата на тъканния фактор на повърхността на тяхната мембрана. Плазмената коагулационна система се активира чрез освобождаване на тъканния фактор в резултат на травма, сепсис, усложнения при бременност, бластемия с хемобластоза.
Системното активиране на коагулацията води до образуване в кръвния поток на прекомерно количество циркулиращ тромбин, което причинява генерализирано образуване на фибрин, активиране и консумация на фактори VIII и V, както и активиране на тромбоцитите. В резултат на това се стартира процесът на масивна микротромбогенеза в микроциркулационната система, водещ до развитието на PON и влошаване на увреждането на ендотелните клетки. От друга страна, генерализираното образуване на фибрин активира фибринолитичната система, фибринолитичните активатори се освобождават активно от активираните ендотелни клетки, тромбоцитите и левкоцитите.
Такава масивна системна активация на хемостаза води до бърза консумация на всички фактори на кръвосъсирването, тромбоцити и инхибитори. Консумацията на тромбоцити в ДВГ може да се извърши изключително бързо, а костният им мозък няма време да попълни циркулиращия пул на тромбоцитите. В допълнение, циркулиращите тромбоцити стават функционално дефектни в резултат на излагане на продуктите на разграждане на фибрина. При чернодробни заболявания намаленият протеинов синтез, дължащ се на чернодробна недостатъчност, допринася за задълбочаващия се дефицит на коагулационните фактори и техните инхибитори. Освен това е важно патологичното разрушаване на коагулационните фактори от бактериални протеази при сепсис, панкреатични ензими при панкреатична некроза или почти фетална течност при емболия на околоплодната течност. Резултатът от тези процеси е кървене, дължащо се на тромбоцитопения, хиперфибринолиза и дефицит на фактор на кръвосъсирването.
КЛИНИЧНИ ПРОЯВИ
Спектърът на клиничните прояви на DIC зависи както от причината, която я причинява (DIC е винаги вторична, не е нозологична форма, а синдром, винаги свързан с голямо заболяване) и състояния, свързани с неговото развитие. При острия ход на ДВС, неговите прояви могат да бъдат признаци на ПОН, което показва увреждане на ЦНС, бъбречна, чернодробна, белодробна дисфункция. Характерни са метаболитна ацидоза, протеинурия, хипоксия, хипотония и треска. Хеморагичните симптоми включват петехии и екхимози на кожата, спонтанно кървене на лигавиците, кървене от места на инжектиране и хирургични рани, при тежки случаи - интрацеребрални кръвоизливи. Микротромбозата причинява исхемично увреждане на вътрешните органи, главно на мозъка, белите дробове, бъбреците и черния дроб. Тромбозата на съдовете на дермата и подкожната тъкан е придружена от акроцианоза, възможното развитие на гангрена на пръстите или краката. Исхемичните признаци на ДВС се проявяват чрез неврологично увреждане на съзнанието (летаргия, бързо изчерпване и едносвързаност при отговаряне на въпроси), хипоксемия и хипоксия с нарушена честота и дихателен ритъм, олигурия или анурия, хипоалбуминемия и хипопротромбинемия.
ДИАГНОСТИКА
Първичната диагноза на DIC се основава изцяло на клиничната картина, включително хеморагични и исхемични признаци. Различават се хипер- и хипокоагулативни фази на остър DIC и хиперкоагулативната фаза на DIC трябва да се диференцира от хроничния хиперкоагулативен синдром (статус), който е фундаментално различен от DIC по отношение на патогенетични и клинични и лабораторни данни.
Хиперкоагулативен синдром - повишена кръвна готовност за съсирване, компенсирана от анти-съсирващи механизми. Когато в съдовата система няма нито локални, нито разпространени кръвни съсиреци, няма клинични прояви на тромбоза. В лабораторни данни обаче се отбелязва скъсяване на АРТТ, протромбиново време, повишена активност на тромбоцитите, намалена фибринолиза, бързо образуване на съсиреци в епруветка.
Хиперкоагулативната фаза на DIC често е преходна и лекарят може да не го диагностицира. Появяват се клинични признаци на органна исхемия. Всички лабораторни признаци на хиперкоагулация (APTT, протромбин, активиране на тромбоцитите) са изразени, но в същото време се появяват първите първоначални признаци на консумация на коагулационен фактор - количеството на тромбоцитите, концентрацията на антитромбин III, протеин С не се намалява рязко. но тя е хлабава и нестабилна. Важен симптом често е бърза тромбоза на игла или катетър по време на интравенозна пункция.
Хипокоагулативната фаза на DIC се характеризира с признаци на дифузна хеморагична диатеза (петехиално-екхимален кръвоизлив) и лабораторни маркери на консумация на коагулационни фактори на хемостазната система - удължаване на времето на кървене, APTT, протромбиново време, значително намаляване на броя на тромбоцитите и техните увреждания. концентрации на фибриноген, фактор VIII, появата на В-димери.
ЛЕЧЕНИЕ
Основата на терапията на ICE е облекчаването на стартиращия патологичен процес. Етиологичната терапия на тези заболявания (антибиотици за сепсис, химиотерапия или хирургична намеса при тумори, плазмафереза \u200b\u200bза остра интраваскуларна хемолиза и др.) Дава ефект само след определен период от време. Ето защо съпътстващите терапевтични мерки са толкова важни, че да осигурят възстановяването и поддържането на BCC, адекватната оксигенация, корекция на хипотонията с цел подобряване на микроциркулацията.
В същото време, критичното състояние на пациента, утежнено от проявите на DIC, налага бърза трансфузионна корекция на нарушения в хемостатичната система.
При лечение на хиперкоагулативната фаза на ДВС, когато има клинични прояви на органна исхемия, дължаща се на микротромбоза, се предписва натриев хепарин. Хепарин натрий инхибира активността на тромбина, като по този начин намалява образуването на фибрин. Обикновено се предписва 8-10 MEDKHH) с непрекъснато интравенозно приложение с дозатор от лекарствени вещества (инфузомат). Трябва да се има предвид, че натриевият хепарин работи ефективно при условие, че има достатъчно количество антитромбин III в кръвната плазма. С намаляването му е необходимо да се преливат FFP (10 ml / kg) или да се предписват търговски препарати от антитромбин III (до 3000 IU / ден). Критерият за ефективността на терапията с хепарин натрий ще бъде намаляване на концентрацията на продуктите от разграждането на фибрин и В-димери, увеличаване на съдържанието на фибриноген, съкращаване на протромбиновото време. Използването на хепарини с ниско молекулно тегло в тази ситуация е непрактично поради липсата на ефективност и невъзможността за мониторинг. При лечението на DIC-синдром на септична етиология, активираният протеин С, който като антикоагулант инхибира активирането на фактори V и VIII и намалява образуването на тромбин, се е препоръчал добре.
Много по-често лекарът от интензивното отделение се среща с хипокоагулаторната фаза на DIC, при трансфузионна терапия, чиято водеща роля принадлежи на недостига на коагулационни фактори. DIC е сложно нарушение на хемостатичната система, следователно „първата цигулка” в нейното лечение се играе от преливане на FFP - комплексна трансфузионна среда, в която оптималният набор съдържа всички необходими коагулационни фактори. Целта на трансфузията на FFP е да се увеличи концентрацията на фибриноген над 1-1,5 g / l. Терапевтичната доза се счита за преливане на FFP в размер на 15-20 ml / kg. При липса на постигане на хемостаза е възможно повторно въвеждане на FFP под лабораторен контрол върху концентрацията на коагулационните фактори. Понякога е необходимо да се добавят препарати с антитромбин III за трансфузия на FFP, особено ако концентрацията му в кръвта е по-малка от 70%. С заплахата от циркулационна претоварване с цел намаляване на обема прибягва до преливане на криопреципитат (една доза на 10 kg телесно тегло). Използването на плазмафереза \u200b\u200bпри лечението на DIC е насочено едновременно към терапията на основното заболяване (например, остър интраваскуларен хемолиза, дължащ се на трансфузия на червени кръвни клетки, които са несъвместими с антигените на ABO системата) и за предотвратяване на циркулаторно претоварване, когато са необходими големи обеми плазма.
Трансфузия на тромбоцити в DIC е показана само с развитието на кървене и намаляване на техния брой по-малко от 50x109 / l. Целта на тромбоцитната трансфузия е да надвиши тази стойност, за която, по правило, е необходима единица тромбоцитен концентрат (55-70x109) на 10 kg телесно тегло за трансфузия. При изразена консумация на тромбоцити са необходими многократни трансфузии на всеки 24 часа.
Трансфузията на червените кръвни клетки се показва само по здравословни причини с потвърдени признаци на хипоксемия и тъканна хипоксия, дължащи се на липсата на червени кръвни клетки в кръвния поток.
Важно е да запомните, че някои пациенти с лабораторни признаци на DIC нямат клинични прояви на тромбоза или склонност към кървене. Такива пациенти не се нуждаят от трансфузионна корекция на хемостаза, те трябва да провеждат терапия за основното заболяване.
СПИСЪК НА ЛИТЕРАТУРА ^
Практическа трансфузиология / Ed. GI Kozinets. - М .: Практика, 2005. - 544 с.
Шевченко Ю.Л., Шабалин В.Н., Заривчатски М.Ф., Селиванов Е.А. Ръководство за обща и клинична трансфузиология. - SPb.: Foliant, 2003. - 608 с.
KN ^ aags! T., Tahapega U.R.K., Войжс! K. e * a1. RNagtasoktellls $ o ^ gesotp: aPu ^ es! ^ asShg VII t 1gayita rayep ^ t1 \\ t Saga. - 2006. - Wo1. 10. - С. 104.
Магистър II., Kepe * S., 5е§а1 Е. е1 а1. KesoshPap! asPua ^ es! Gus ^ i VII bf афиширане на ltoggagage sopt1t 1t; походка //]. Tgaita. - 2001. - Wo1. 51. - С. 431-438.
Упр .:! .B., Ko $$ am1: K., Cui V. e * a1. Kesottepsa1: Да $ op 1bе и $ e за! "Hesottap! Asy-ua * es1 ^ as1; og VII az a af" ipsuye 1; gea1: tep1; Gogta \u200b\u200b$$ 1уе ееес11п§ - eеorep regresresIue // Cn. (Sag. - 2006. - Wo1. 10. - стр. 120.
Пациент с скорбут има кървене на венците и петехии върху кожата. Какво причинява нарушения на хемостазата при това заболяване?
А. @ Нарушаване на синтеза на колаген
B. Тромбоцитопения
В. Излишък от антикоагуланти
D. Активиране на фибринолизата
Д. Прокоагулантна недостатъчност
Момчето с хеморагичен синдром няма антигемофилен глобулин А в кръвта (фактор VIII). Какъв е механизмът на неуспех на хемокоагулацията при този пациент?
Вътрешен механизъм на образуване на протромбиназа
Превръщането на фибриноген в фибрин
Външен механизъм на образуване на протромбиназа
Превръщането на протромбин в тромбин
Втягане на кръвен съсирек
Човек, който е страдал от хронична миелоидна левкемия в продължение на две години, е приет в болница в състояние на остра бъбречна недостатъчност. Каква може да е причината за остра бъбречна недостатъчност при този пациент?
@DIC - синдром
лимфопения
неутропения
тромбоцитопения
Недостатъчната активност на коагулационните фактори причинява развитието на хеморагичен синдром при пациенти с хиповитаминоза К?
@ X, IX, VII, II
Факторът на Willebrand
Пациент, който системно консумира ацетилсалицилова киселина, разви кръвоизлив. С намаляването на активността на които тромбоцитните ензими се свързват с развитието на тромбоцитопатия в този случай?
Циклооксигеназа
липоксигеназа
пероксидаза
Цитохромоксидаза
Глюкоза-6-фосфат дехидрогеназа
Момичето периодично има кървене от носа, има малки хеморагични обриви по кожата. Проучването установи: време на кървене - 10 минути, намалена адхезивна тромбоцитна способност и ниска активност f. VIII (VIII: С). Каква е болестта при дете?
@ Von Willebrand болест
Хемофилия А
Хемофилия В
Наследствена дисфибриногенемия
тромбоцитопения
След страдание от ангина, 5-годишно момиче има петехиален обрив по кожата на торса и крайниците, кървене от венците. Изследването показва намаляване на броя на тромбоцитите в кръвта. На какво ниво на тромбоцитопения (g / l) се появяват клиничните му признаци?
Какво може да бъде основната връзка в патогенезата на тромбофилията при пациент, страдащ от тромбофлебит на долните крайници?
@ Липса на антикоагуланти
тромбоцитопения
thrombocytopathy
Прокоагулантна недостатъчност
За целите на диагностицирането на каква промяна в хемокоагулационната система е предписано изследване на нивото на продуктите от разграждането на фибрина в кръвта?
thrombocytopathy
тромбоцитопения
Вазопатия хеморагична
Пациентът е диагностициран с генетичен дефект в мембранния рецептор за фактор на von Willebrand - гликопротеин (GP) Ib, който е отговорен за началния етап на адхезия на тромбоцитите към колагена. Какво е името на това заболяване?
@ Bernard - Soulier
Фон Willebrand
Адисън - Бирмера
Уилсън - Коновалова
Гланцмана - Негели
Пациентът е диагностициран с наследствен дефект на GP IIb - IIIa - мембранен рецептор, който осигурява свързването на фибрин с тромбоцитната мембрана и е необходим за тяхното агрегиране. За какво става дума?
@Glancman - Негели
Фон Willebrand
Адисън - Бирмера
Уилсън - Коновалова
Бернар - Сулие
При едно дете с комбиниран имунен дефицит кръвен тест показва намаляване на адхезията на тромбоцитите към колагена, тяхното агрегиране, отслабване на кръвосъсирването и ретракция на кръвни съсиреци. При кои имунодефицити се наблюдават подобни промени?
@ Whiskott - Aldrich
Швейцарски тип
Ди - Джорджи
Nezelofa
Болно момиче е диагностицирано с тромбастения на Гланцман. Какво нарушение в системата на хемостаза се случва в този случай?
@ Дизагрегационна тромбоцитопатия
Абсолютна тромбоцитопения
Дисперсивна тромбоцитопатия
Недостатъчна тромбоцитопатия
Дистгранулационна тромбоцитопатия
Членовете на експедицията, които са били на север, се оплакват от кървене от венците и петехиални кръвоизливи по кожата. От анамнезата е известно - в храната не е имало достатъчно аскорбинова киселина и това е довело до крехкост на съдовата стена. Каква е патогенезата на вазопатията?
@ Диспластика
възпалителен
метапластични
дистрофията на
с имунитет
При пациент след продължителна операция на панкреаса, следоперативната рана е кървяла дълго време. Според коагулограмата е установено значително увеличение на нивото на плазмин. Какво е патогенезата на коагулопатията, наблюдавана в този случай?
@ Фибринопатия
Protrombinazopatiya
Tromboplastinopatiya
Trombinopatiya
Vazopatiya
Пациентът е диагностициран с генетичен дефект на фактор V, който става нечувствителен към инактивиране от антитромботичния комплекс тромбомодулин - протеин С, който намалява способността на съдовата стена да ограничава образуването на фибрин. Каква патология на кръвосъсирването ще възникне с тази аномалия?
@ тромбофилия
тромбоцитопения
thrombocytopathy
Хеморагичен синдром
Комплексът от механизми, поддържащи кръвта в течно състояние, без неговата коагулация в лумена на съда или изтичане през съдовата стена, се нарича хемостаза. Тъй като патологичните състояния, свързани с коагулацията на кръвта, са разгледани в следващата лекция, тук трябва да се обсъдят други промени, свързани с нарушена хемостаза.
Кървене (кръвоизлив, от гръцки. Haima - кръв и рейн - поток) е притока на кръв извън съдовото легло или сърцето в околната среда (външно кървене) или в кухината на тялото, лумена на кухия орган (вътрешно кървене). Примери за външно кървене са метрорагия (матка), мелена (чревна) и вътрешно кървене - хемоперикард, хемоторакс, хемоперитонеум и хемартроза (съответно в кухината на перикарда, плеврата, коремната кухина или ставата).
В зависимост от източника на кървене, те се разделят на артериални, венозни, артериално-венозни (смесени), капилярни, паренхимни (капилярни от паренхимни органи) и сърдечни.
Особен вид кървене е кръвоизлив, при който кръвта се натрупва екстраваскуларно в тъканите. Има четири вида:
Хематом - хеморагия с нарушена целостност на тъканта и образуване на кухини;
Хеморагично накисване (инфилтрация) - кръвоизлив с запазване на целостта на тъканта;
Синина (синина) - планов кръвоизлив в кожата, подкожната тъкан, лигавиците;
Petechiae - кръвоизливи в кожата, лигавиците и серозните мембрани, вътрешните органи.
Множество петехиални кръвоизливи, сливащи се в по-големи, се наричат \u200b\u200bхеморагична пурпура, а синината до 2 см в диаметър се нарича екхимоза.
Механизмите на развитие на кървене и кръвоизлив включват:
Разкъсване (хеморагия на рексин) в резултат на травми на непроменен съд или некроза (разкъсване на сърцето при инфаркт на миокарда), възпаление (сифилитичен мезаортит с руптура на аортата), аневризма на засегнатата съдова стена;
Корозия (haemorragia на diabrosin), или arrosive кървене, което се развива в разрушаването на възпалението на съдовата стена (по-гноен), злокачествен тумор, некроза (случаен некроза кървене туберкулозен кухина), излагане на химикали (стомашен сок може да доведе до кървене от стомашни язви), покълване вълни на хорионните съдове на маточната тръба при ектопична бременност;
Диапедезис (haemorragia per diapedesin, от гръцки. Dia - през, pedao - скачане), характеризиращ се с кръвна продукция поради повишена съдова пропускливост, като правило, непокътнати съдове на микроваскулатура при тежка хипоксия, интоксикация, инфекция, различни коагулопатии, хеморагична диатеза. Сравнително често диапедичните кръвоизливи се развиват при хипертонична криза, системни васкулити, левкемия, хемофилия, уремия.
Тенденцията към спонтанно кървене или кръвоизлив в отговор на дори незначителни увреждания се нарича хеморагична диатеза. Това състояние е свързано с качествени или количествени промени в тромбоцитите, недостатъчност на хемокоагулацията, патологична чупливост или повишена пропускливост на съдовата стена, както наследствена, така и придобита.
Основните причини за кървене (кръвоизлив) са:
1. Повишена чупливост на съдовата стена се забелязва в неговите вродени дефекти, инфекции и интоксикации, хипо- и бери-бери, стероидна терапия;
2. Тромбоцитни дефекти. Те включват тромбоцитопения от всякакъв произход, наследствено или придобито отслабване на адхезията (болест на von Willebrand, Bernard-Soulier, наследствена хеморагична диатеза и др.), Отслабена агрегация или намаляване на тромбоцитната секреция;
3. Недостатъчност на вродени фактори на кръвосъсирването (хемофилия А по фактор VIII, хемофилия В по фактор IX, болест на фон Вилебранд, други фактори на кръвосъсирването) или по-често с придобит произход (с чернодробни заболявания, дефицит на витамин К, някои имунни лезии);
4. Превишена интраваскуларна коагулация, например при дисеминирана интраваскуларна коагулация на кръвта - DIC (виж следващата лекция).
Резултатът от хеморагии може да бъде благоприятен (резорбция на изтичащата се кръв, организация, капсулиране, образуване на "ръждясала" киста) и неблагоприятна (нагряване, когато инфекцията е прикрепена).
Стойността на кървенето се дължи на външния му вид, тежест и продължителност. Така разкъсването на сърцето при инфаркт на миокарда с образуването на хемоперикард бързо води до смърт на пациента, въпреки че общото количество на кървене обикновено е не повече от 100-200 г. При артериално кървене може да се развие масивна загуба на кръв и остра анемия с фатален изход. Продължителното леко кървене от хронична стомашна или дуоденална язва причинява хронична постхеморагична анемия. Стойността на кръвоизливането зависи преди всичко от неговата локализация и едва след това от нейната големина. Дори малък кръвоизлив в мозъка може да причини увреждане на жизнените центрове, подуване на мозъка и смърт на пациента, докато дори масовите неусложнени кръвоизливи в подкожната тъкан не представляват опасност за живота.
Плазморагията е излизане от лумена на съда с кръвна плазма с импрегниране на околните тъкани (плазмено накисване) поради повишена съдова пропускливост. Плазморагията се появява трансендотелиално поради ултрафилтрация (плазмен изход през порите на базалната мембрана на ендотелиума поради увеличаване на хидростатичното или осмотичното налягане), дифузия (поради градиента на плазмените компоненти в лумена и извън съда), микровезикуларен транспорт (микропиноцитоза или цитотомична система). , Възможно е и интер-ендотелно плазмено освобождаване. Така, плазморагията се определя от увреждане на васкуларната стена (предимно неговата интима) и промяната в кръвните константи. Морфологичното изследване на съдовата стена на микроциркулационното легло се сгъстява, става хомогенно и при електронно микроскопско изследване в подутите ендотелни клетки се наблюдават голям брой микровезикули, образуването на фенестри и тунели, появата на междуклетъчни пукнатини, разхлабване на мембраната на интимата. Натрупването на плазмени компоненти води до увреждане на клетките и междуклетъчно вещество в съдовата стена и периваскуларните тъкани, в края на които се развива хиалиноза, а в тежки случаи - фибриноидна некроза.
Шокът е тежко патологично състояние, характеризиращо се с кръвоносен колапс (остра недостатъчност на кръвообращението) след свръхсилно излагане на хемостаза. Различават се хиповолемични, сърдечни, септични и съдови типове шок.
Хиповолемичният шок се причинява от бързо намаляване на обема на циркулиращата кръв с 20% или повече, което се наблюдава при остра загуба на кръв и дехидратация. По този начин загубата на течности и електролити е възможна при големи изгаряния (дължащи се на освобождаване на плазмата от увредена микроваскулатура), с тежко повръщане, обилна диария.
Сърдечният шок се развива в отговор на намаляване на инсултния обем с кардиологични лезии, наблюдавани при миокарден инфаркт, тежък миокардит, остра митрална или аортна недостатъчност, тромбоза на протезна клапа, руптура на интервентрикуларната преграда, хемотамптон на сърдечната риза. Явно спадане на кръвното налягане води до значително намаляване на кръвоснабдяването на тъканите, подобно на хиповоемичните промени.
Септичен (токсично-инфекциозен) шок се появява, когато има инфекция, причинена от грам-отрицателни (Е. coli, Proteus, Klebsiella и др.), По-рядко грамположителни (стафило-, стрепто-, пневмококи) микрофлора. Екскретираните токсини (предимно ендотоксини) активират комплементната система, коагулацията, фибринолизата, както и тромбоцитите и неутрофилите. В резултат на това се стимулира образуването на азотен оксид (мощен вазодилататор), туморен некрозисен фактор а и интерлевкини, причиняващи остра циркулаторна недостатъчност.
Съдовият (редистрибутивен) шок може да бъде неврогенен (травматичен, болезнен, с увреждане на гръбначния мозък, като усложнение на анестезията) или анафилактичен, причинен от генерализирани реакции на свръхчувствителност. Поради силно изразената вазодилатация, повишената капилярна пропускливост и артериовенозното отделяне, вътрешносъдовият кръвен обем се преразпределя, придружен от значително намаляване на общата периферна съдова резистентност.
Шокът в неговото развитие преминава през три етапа:
1. Непрогресивният (ранен) стадий на шок се характеризира с намаляване на кръвното налягане и сърдечния дебит, като същевременно се запазват жизненоважни органи с относително нормално кръвоснабдяване. Това се дължи на компенсаторната вазоконстрикция на съдовете, особено на кожата и червата. С изчерпването на адаптивните механизми, шокът преминава в следващия етап;
2. Прогресивният стадий на шока се характеризира с изразени клинични симптоми, дълбок колапс, дължащ се на ниско кръвоснабдяване на всички органи и тъкани (тъканна хипоперфузия поради увеличаване на артериалната дилатация), развитие на метаболитни и циркулаторни нарушения. 3. В необратимия стадий на шока има изразена циркулаторна недостатъчност на нивото на микроваскулатурата с нарушение на целостта на съдовата стена, бързо увеличаване на мултиорганната недостатъчност, водеща до смърт на пациента.
Морфологичното изследване показва генерализирани дистрофични и некротични промени, явления на DIC синдром (петехиални кръвоизливи, стазис, кръвни съсиреци в микроваскулатурата). В допълнение, поради особеностите на структурата и функционирането на различните органи, във всяка от тях възникват особени промени - шокови органи. По този начин развитието на некротична нефроза (некроза на сложен епител на тубулите) е характерно за шоков бъбрек. Шоковият бял дроб се проявява с огнища на ателектазата, серозен хеморагичен оток, понякога със загуба на фибринови филаменти (хиалинови мембрани). В мозъка настъпва исхемична енцефалопатия, която се проявява чрез оток, точкови кръвоизливи и огнища на некроза. В сърцето се наблюдават малки, предимно субендокардиални огнища на кръвоизлив и некроза на миокарда, мастна дегенерация на кардиомиоцити с признаци на тяхното ретракция. В кортикалния слой на надбъбречните жлези се наблюдава намаляване до пълното изчезване на липидите, използвани за синтеза на стероидни хормони. В стомашно-чревния тракт се откриват хеморагии, ерозии и остри язви в лигавицата. Шоковият чернодробен шок се отличава с мастна дистрофия на хепатоцитите, а в някои случаи дори и от тяхната центролобуларна некроза.
Прогнозата за шок зависи от неговия тип, тежест, стадия на започване на лечението, наличието на усложнения. Понастоящем при тежък кардиогенен или септичен шок смъртността достига 50% или повече.
Хемостатични нарушения
Комплекс от фактори и механизми, които осигуряват оптимално агрегатно състояние на агрегатното състояние на кръвта.
За оценка на системата за кръвосъсирване се извършват тестове:
Време, продължителност на кървенето. Пробийте пръст и стърчащ капка, потопен в лист хартия. Норма - 45-180 секунди.
Времето за съсирване според Lee-White. Взема се в епруветка и се люлее до появата на фибринови влакна. Норма - 6-9 минути.
В отговор на васкуларно увреждане настъпва спазъм на съдовата стена. Тромбоцитите бързат към увредения съд, прилепват към стената на съда (адхезия). Тромбоцитният тромб се слепва и образува. Тези механизми са съдова тромбоцитна хемостаза.
Фазова коагулация.
Коагулационна хемостаза. Активирането му може да се осъществи по два начина:
Механизмът е вътрешен, активира се с помощта на колаген, протеаза или адреналин, тези фактори водят до факта, че: XII фактор Hageman се превръща в активен протеин, когато се активира (аминокиселинната група пада и е активна форма)) XIIa a XI IX VIII - задължително с участието на калиеви йони формиране на активна протромбиназа (тромбопластин).
Механизъм - външно - тъканно увреждане. Тютюневият тромбопластин (III) допринася за прехода на фактор VII в активната форма и резултатът е образуването на активна протромбиназа.
Във фаза 1 на кръвосъсирването участват 7, 8, 9 коагулационни фактора.
Съсирване на кръвта
Активната протромбиназа (тромбокиназа) се превръща в тромбин. Той има 2 под-фази:
Образуване на ензимна доза от тромбин.
Под действието на дозата на изпичане на тромбин, проконвертин (VII) преминава към конвертин (VIIa). Convertin връща процеса на коагулация във фаза 1
Проацелирин (V) отива към Accelerin (Va) - ускорява кръвосъсирването.
Образуване на получената доза тромбин. Работа протромбин, проакцелин, проконвертин.
Тромбинът действа върху фибрина, образувайки фибриноген. Полимеризация и стабилизация. Фибриноген - фибринономер - фибринови олигомери - фибринполимери.
Фаза - фибринолиза.
XII II XIIa min плазминоген - плазмин (основният фактор на фибринолитичната система) фибрин се разпада до продукти от разграждане на фибрин (продуктите на фибринолиза активират коагулацията от фаза 1).
Система за хемостаза.
Хеморагична диатеза.
Разделят се според компонента на увреждане на хемостазата:
Vazopatii:
наследствен:
телеангиектазия или болест на Рандюс-Ослер - автозомно доминантно наследство, т.е. дефект в структурата на съдовата стена, става по-тънък и се образуват подобни на торби ектази. Локализация: устни, горната част на торса, лицето, скалпа, лигавицата, кухините. Тези изтънени стени са добре наранени. Пациентът има хронична желязодефицитна анемия поради хронична загуба на кръв.
придобит:
Хиповитаминоза С (скорбут), РР - необходими за полимеризацията на съдовата стена - пациентът има петехии, васкулит. На симетрична кожа.
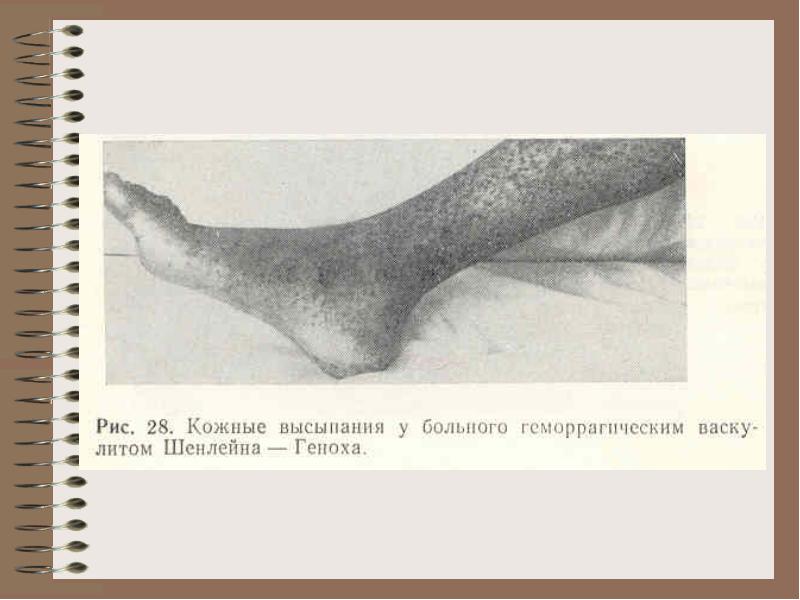
Хеморагичен васкулит (болест на Schönlein-Genoch):
Проби от съдовата стена съпротива: Konchalovsky (налягане в маншета е под налягане до определена стойност, след това броят на петехиите се брои), турникет (след прилагане на маншет, когато измерване на кръвното налягане, гледане на кожата - ще има кръвоизливи), прищипване (прищипване на кожата).
thrombocytopathia:
тромбоцитопения:
Болестта на Verlgof (тромбоцитопенична пурпура) се наследява автозомно доминантно. Проявява се под формата на синини и цъфтят - кожата на леопард.
Способността на далака да произвежда инхибитор на тромбоцитопоезата (далачен фактор) се наследява. Този фактор инхибира оттеглянето на тромбоцитите от мегакариоцитите в костния мозък, броят на мегакариоцито се увеличава и няма смисъл от тях.
Лекувайте: отстраняване на далака.
Придобит: НА ТЕЛЕФОН ФОТО!
Миелотоксични ефекти:
Екзогенни фактори: физични (йонизиращи лъчения), химически (цитостатици, антибиотици, рафинирани продукти), биологични (вируси, токсини от микроорганизми).
ендогенната:
Потискане на нормалните кълнове на кръвообращението.
Автоимунна тромбоцитопения.
Резултатът - назално, гингивално кървене, матка, синини.
За диференциация има биоанализ, серумът на пациента се инжектира в животното. Слендовият фактор няма видова специфичност, поради което причинява тромбоцитопения при животно.
Тромбоцити - нарушение на функционалните свойства на тромбоцитите:
Наследствена: болест на Гланцман, болест на Willebrand (сложна хеморагична диатеза, тъй като ендотелът обикновено произвежда фактор на Willebrand, компонент 8 на фактора, а изкривеният синтез на този фактор е наследен. Факторът Willebrande е коагулационен фактор).
придобит:
Преливане на големи дози кръв, плазма, прокоагулантни концентрати.
Миелом, болест на Waldenstrom (увеличен в пика на нормалните и абнормните протеини).
DVSS (увеличен PDF - продукти от разграждане на фибрина)
Лекарствени - НСПВС
Хиповитаминоза С, В12.
Механизъм на тромбоцитоза:
Нарушаване на синтеза и натрупване в гранулоцити на тромбоцитни биологично активни вещества,
Нарушаване на дегранулация и освобождаване на тромбоцитни фактори в кръвната плазма.
Диагностика на тромбоцитопения и астения:
Броят на тромбоцитите: 180-400 * 10 9 / l.
Време за агрегиране на тромбоцитите: 14-18 секунди
Процентът на клетките, които са се присъединили към агрегацията, е повече от 90.
Удължаване на времето и намаляване на клетките, които са влезли - тромбоцистин.
Положителният тест на Кончаловски - тромбоцитите определят трофичността на съдовия ендотелиум, излъчват растежни фактори, необходими за ендотелиоцитите. Ако тромбоцитите са малки - дефекти, нестабилността на съдовете се увеличава.
Коагулопатия - нарушение на коагулационната хемостаза:
Наследствен. Във фази на коагулация:
Разрушаване на фаза I на коагулацията:
Хемофилия А (80% сех хемофилия) е дефицит на фактор VIII.
Хемофилия В (10%) е дефицит на фактор IX.
Хемофилия С (5%) - дефицит на фактор XI.
Механизмът на наследяване на хемофилия: хемофилия А е дефект на Х-хромозомата. Здрав баща, майка - превозвач: здрав син, здрава дъщеря, дъщеря-превозвач, син, страдащ от хемофилия. Болестта се проявява веднага след раждането, новороденото може да има различни кръвоизливи (цефалгематома). Всяко нараняване води до образуване на хематоми (вид кръвоизлив). Може би образуването на хемартроза.
B и C хемофилия може да се прояви както при мъже, така и при жени.
Диагностика на нарушения на фаза I коагулация:
Активирано време на плазмената рекалцификация (AVR) 40-60s.
Частично активирано тромбопластиново време (APTT) 33-45 сек.
Фаза II Прекъсване:
Наследствена (парахемофилия): хипопротромбинемия, хипопроконвернемия, хипопроцелеринемия.
Придобити: чернодробна недостатъчност, ахолия, хиповитаминоза К.
За оценка на състоянието на фаза II, ние оценяваме протромбиновия индекс (PTI) от 85-110%.
Фаза III Прекъсване:
Наследствена: хипофибриногенемия, афибриногенемия, дисфибриногенемия.
Придобити в резултат или на намаляване на синтеза, или на повишена консумация (особено при DVSS): хипофибриногенемия, афибриногенемия.

ПАТОФИЗИОЛОГИЯ НА СИСТЕМАТА НА ХЕМОСТАЗА
Стойността на хемостатичната система
1. Запазване на кръвта в течно състояние (адекватно съотношение на активността на коагулационните и антикоагулационните системи) \\ t
2. Предотвратяване и спиране на кървенето (поддържане на постоянен обем на циркулиращата кръв)

ВИДОВЕ ХЕМОСТАЗ
Съдова-тромбоцитите
(Първоначално)
- СТОПВАНЕ НА КРЪЩЕНЕ В МИКРО КОРАБИ
коагулацията
(Вторичен)
ФОРМИРАНЕ НА ФИБРИНОВИ ПАКЕТИ

КОМПОНЕНТИ НА ХЕМОСТАСТИЧНАТА СИСТЕМА
* Васкулна стена
* Тромбоцитите (моноцити, еритроцити)
* Плазмени системи:
- PROKOAGULYANTY
- АНТИКОАГУЛАНТИ
- FIBRINOLYTIC
- KALLIKREIN-KININOVAYA

КЛАСИФИКАЦИЯ НА РАЗЛИКИ НА ХЕМОСТАЗА
ЗА ЕТИОЛОГИЯ
- ХЕРДИТАРНИ
- Придобита
ЗА МЕХАНИЗМА НА РАЗВИТИЕТО
- Васкуларни разстройства
хемостаза
- ВЪЗСТАНОВЯВАНИЯ НА КОАГУЛАТОРНИЯ ХЕМОСТАС
ЗА НАПРАВЛЕНИЕТО НА ПРОМЕНИТЕ
- Хипокоагулация
- ХИПЕРКОАГУЛИРАНЕ

gipokoagulyatsii
Намалена способност на кръвта да се съсирва с тенденция към повторно кървене и кръвоизлив (спонтанни или след леки наранявания)

етиология
1. ТРОМБОЦИТОПИЯ
2. тромбоцитопатии
3. VASOPATHY
4. КОАГУЛОПАТИЯ

тромбоцитопения
Патологично състояние, характеризиращо се с намаляване на броя на тромбоцитите в кръвта. (по-малко от 150 · 109 / l)

НАСЛЕДСТВЕНА ТРОМБОЦИТОПЕНИЯ
По правило това е едновременно съпроводено с анормални тромбоцитни дефекти

ПРИДОБИВАНА ТРОМБОЦИТОПЕНИЯ (КЛАСИФИКАЦИЯ ПО МЕХАНИЗМА НА РАЗВИТИЕТО)
УВРЕЖДАНЕ НА ПЛАТЕЛИТЕ
- имунни комплекси
- механична травма (спленомегалия, хемангиома)
ЗАКЛЮЧЕНО ФОРМИРАНЕ НА ПЛАТЕЛИТЕ
(апластична анемия, химическо и радиационно увреждане на червения костен мозък, замяна на хематопоетична тъкан с тумор)
УВЕЛИЧЕНО ИЗПОЛЗВАНЕ НА ПЛАТЕЛИТЕ
(тромбоза, DIC)

ИММУННА ТРОМБОЦИТОПИЯ
GETEROIMMUNNAYA
* По-често се среща в деца
** причина - промени в антигенната структура на тромбоцитите (по време на слягането на рубеола, едрата шарка, аденовирусните вируси; лекарствените хаптени - хинидин, сулфонамиди, рифампицин; ваксини)
***Благоприятен курс (ако причината е елиминирана, възниква пълно възстановяване)

ИММУННА ТРОМБОЦИТОПИЯ
АВТОИМУНЕН
По-често се среща възрастни
причина - липса на имунен толеранс към антигени на собствените тромбоцити
Провокативни фактори: лекарства, вируси, бактерии

Автоимунна тромбоцитопения
БОЛЕСТ НА ВЕРЛОГОФ
(автоимунна хронична тромбоцитопенична пурпура)
* На броя на повърхността на тромбоцитите Ig G увеличава 10 пъти
Основното място на синтеза на Ig G е далак
* Принцип на лечение:
- спленектомия
- кортикостероиди
- имуносупресори
* Няма пълно излекуване.

thrombocytopathy
Нарушена хемостаза поради качествена малоценност или дисфункция на тромбоцитите, която се характеризира с нарушение на съдова тромбоцитна хемостаза, поява на кървене на тъканите и органите \\ t

БЕЗ НАРУШЕНИЕ НА ОТГОВАРЯНЕТО НА ОТГОВОРНОСТТА НА ГРАНУЛИ
Гланцман Тромбастения
* Наследяване - автозомно рецесивен
*причина - отсъствието на гликопротеини 2Ь и 3а в черупката на тромбоцитите
*патогенеза- тромбоцитите не взаимодействат с фибриногена и не се агрегират
*Симптоми: петехии, кървене от носа, кървене от матката ( може да бъде смъртоносно !!)

Наследствена тромбоцитопатия
С НАРУШЕНИЕТО НА ОТГОВАРЯНЕТО НА ОТГОВОРНОСТТА НА ГРАНУЛИ
наследяване - автозомно рецесивен
причина - нарушена циклоксигеназна активност, ниска контрактилна протеинова активност
патогенеза - липса на агрегация при взаимодействие с колаген, липса на освобождаване на гранули
Симптоми:

Наследствена тромбоцитопатия
С НАРУШАВАНЕ И ОСВОБОЖДАВАНЕ НА СЪДЪРЖАНИЕТО НА ГРАНУЛИ
Болест на Херман-Пудлък (AR)
* причина - нарушение на натрупването на плътни гранули (АДФ, адреналин, серотонин, Са2 +)
* патогенеза - при взаимодействието с колаген няма агрегиране, не се освобождава съдържанието на гранулите
* Симптоми: петехии, кървене от носа, кървене от матката

Наследствена тромбоцитопатия
С УВРЕЖДАНЕ НА ЛЕЧЕНИЕ И АГРЕГИРАНЕ НА ПЛАТЕЛИТЕ
Синдромът на Villebrand-Jurgens (AR)
причина - Дефицит на фактор Willebrand
патогенеза - нарушена е адхезията на тромбоцитите, дължаща се на дефицит на фактор 8
Болест на Bernard Sul (AR)
причина - липса на гликопротеин 1 върху тромбоцитите
патогенеза - взаимодействието на тромбоцитите с фактора на von Willebrand, f. 5, f. 11
Признаци на - капилярно кървене ( особено опасен по време на пубертета или раждането

Наследствена тромбоцитопатия
НЕДОСТАТЪЦИ И НАМАЛЕНА НАЛИЧНОСТ f.3
Тромбоцитопатия Боу и Овен
причина - дефицит f.3 тромбоцити
патогенеза - няма взаимодействие на тромбоцитите и прокоагулантите
Симптоми: петехии, кървене от носа, кървене от матката

Наследствена тромбоцитопатия
Тромбоцитопатия, комбинирана с други наследствени аномалии
Синдром на Whiskott-Aldridge
- причина - в тромбоцитите има няколко плътни гранули (ADP, серотонин, адреналин, Ca2 +), алфа гранули (бета-тромбоглобулин, фибриноген, фибронектин, растежен фактор)
- патогенеза - Намалена адхезия и агрегация на тромбоцити, нарушено освобождаване на гранулите
- Симптоми:хеморагичен синдром се появява рано, може да има фатален кръвоизлив

Придобита тромбоцитопатия (етиология)
1. левкемия- малко гранули в тромбоцитите, поради ускореното съзряване, намалена адхезия и агрегация
2. НатрупванеIg M - увреждане на рецепторите с имунни комплекси, нарушение на взаимодействието на тромбоцити с прокоагуланти (имунни заболявания)
3. хиповитаминоза В12 - разрушено освобождаване на гранули
4. Ефекти на лекарството

Тромбоцитопатия на лекарството
* И инхибитори на синтеза на тромбоксан А2
стероидни противовъзпалителни средства
- нестероидни противовъзпалителни средства (аспирин блокира тромбоцитната агрегация за 4-6 дни)
* Образователни стимулатори сАМР
-papaverin
- еуфилин
-анаболични стероиди
* Саон-антагонисти
-verapamil
-korinfar

VAZOPATIYA
Хеморагична диатеза поради функционална и морфологична малоценност на съдовата стена
- вродена
- придобити

HORN VASOPATHY
Бол. Randyou-Osler (хеморагична телеангиектазия)
Бол. Фабри (дифузен ангиокер-тома на ствола)
Наследствена тромбоцитопенична микроангиоматоза

HORN VASOPATHY
причина - наследствено нарушение на развитието на съединителната тъкан, включително васкуларен субендотелиум
особеност
- фокално съдово изтъняване
- разширяването на лумена на микроспитите
- малко колагенови влакна в субендотелиума
- съдовете лесно се нараняват
- лоша адхезия и агрегация на тромбоцитите, поради липса на колагенови влакна
**Признаци на - кървене на носа, белодробно-бронхиален и стомашно-чревен тракт (има фатален изход)

ПРИЕМА ВАСОПАТИ
1. И diopaticheskaya (Сарком на Капоши)
- етиология - неизвестна
2. застоял (дерматит Klotza, дерматит Favra Rakusho)
- етиология - хронична сърдечна недостатъчност, локална венозна недостатъчност
3. дистрофията на
Стероидна пурпура - хиперфункция на надбъбречните жлези, лечение с кортикостероиди - инхибира синтеза на колаген
Изгаряне - дефицит на Vit.S
Bol.Shenlein-Genokha - съдово увреждане от имунни комплекси
4. неврогенен
Клинични признаци - кожни форми на кървене

коагулопатия
Хеморагична диатеза, която възниква в резултат на патологията на коагулационната система на хемостазата
** наследствен
** придобити

Наследствена коагулопатия
Генетично причинено нарушение на коагулацията поради дефицит или молекулярна аномалия на вещества, отговорни за коагулационната хемостаза

Наследствена коагулопатия
КЛАСИРАНЕ
1. Коагулопатия, дължаща се на изолирано нарушение на вътрешния механизъм на образуване на протромбиназна активност (хемофилия А, В, С, Willebrand b., Hageman дефицит) \\ t
2. Коагулопатия, дължаща се на изолирано нарушение на външния механизъм на образуване на протромбиназна активност (хипопроконвернемия - дефицит от 7 л.).
3. Комбинираното нарушение на външния и вътрешния механизъм на образуване на протромбиназна активност (парахемофилия - дефицит от 5 lb., b.Steuart-Prouer - дефицит от 10 lb.)
4. Нарушаване на крайния етап на кръвосъсирването (афибриногенемия)

СТАТИСТИКА
Сред всички форми на коагулопатия страдат:
Хемофилия А 68 - 78%
B , Willebrand 9 - 18%
Хемофилия В 6 - 13%
Хемофилия С, парахемофилия и хипопроконвернемия 1 - 2%
Останалите форми - клинична казуистика

Хемофилия А
Хеморагична диатеза поради наследствен дефицит на прокоагулантната част на фактор 8
Фактор 8 (протеин с високо молекулно тегло)
1. Гликопротеин прокоагулант (VIII: K)
2. Гликопротеин, осъществяващ адхезия на тромбоцити (VIII: FV)
3. Гликопротеин активираща адхезия на тромбоцити под влиянието на ристомицин (VIII: Rkoff)
4. Антигенен маркер VIII: K (VIII: K AG)
5. Антигенен маркер VIII: Rkof (VIII: Rkof AG)
Дейност VIII: K и VIII: PV намалява с намаляване на мултимерната структура и само 8 фактора

Хемофилия А
* tiologiya - генна аномалия в X хромозомата, която контролира синтеза прокоагулантна част f. 8 (Viii: k)
** Ill - мъже (46, XhY)
** видове
- синтезира се хемофилия А + (антиген-положителна форма - аномална) VIII: К), 8-10% страдат
- Хемофилия А- (антиген-отрицателна форма - не синтезирана) VIII: К), страдат 90–92%
**** клиника: кръвоизливи в големи стави, хематоми (подкожно, интрамускулно), тежко и продължително посттравматично кървене. Възможно кървене в коремните органи, стомашно-чревно кървене

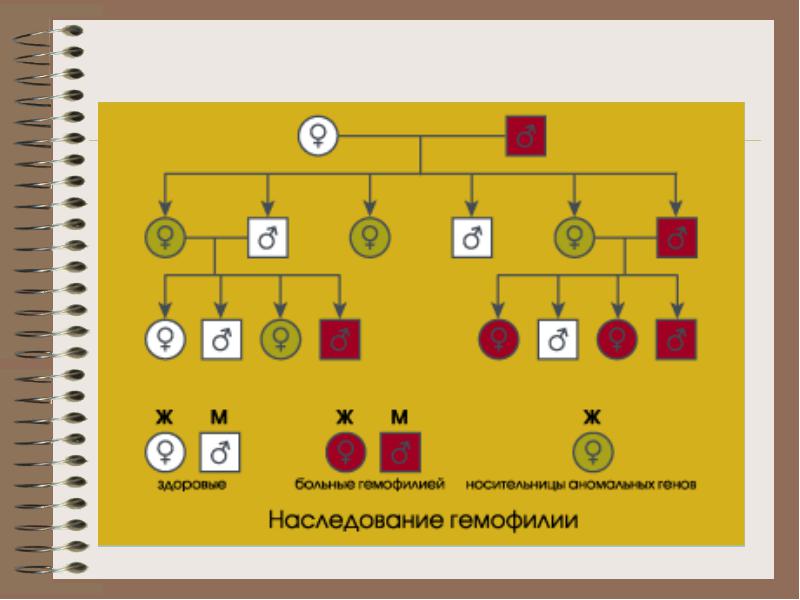
хемофилия
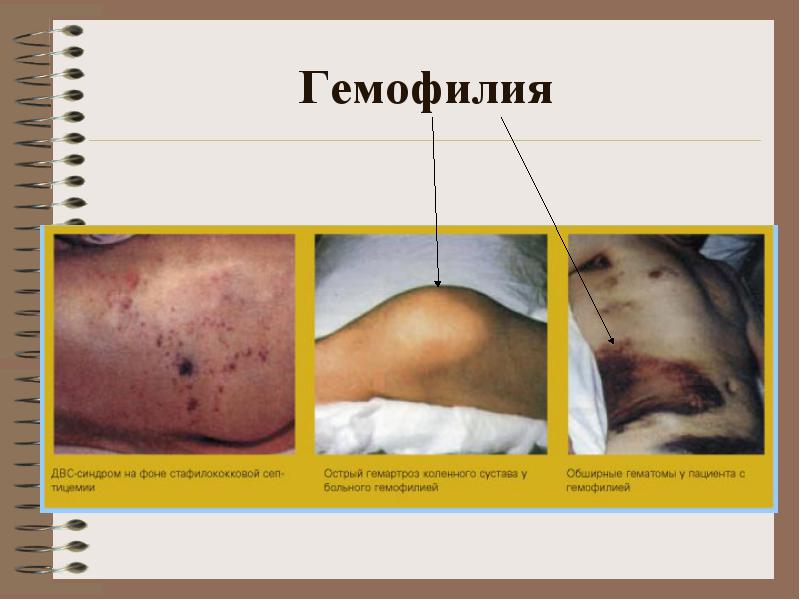
хемофилия
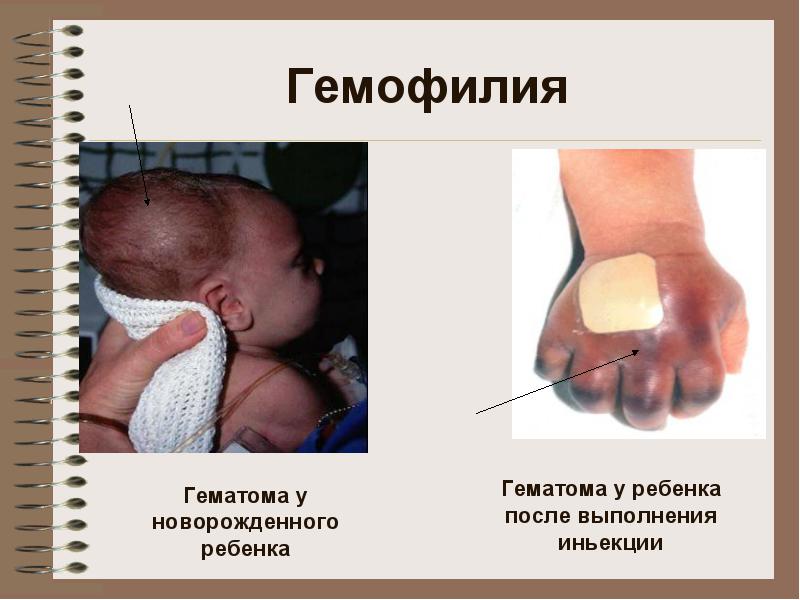
Хемофилия В
етиология - генна аномалия в X хромозомата, която контролира синтеза на f. 9
Болни - мъже (46, XhY)
- жени (46, XhXh), (45, Xh O)
*** Изгледи
- синтезира се хемофилия В + (антиген-положителна форма - аномална) е. 9)
- Хемофилия В- (антиген-отрицателна форма - не синтезирана) е. 9)
клиника: кръвоизливи в големи стави, хематоми (подкожно, интрамускулно), тежко и продължително посттравматично кървене. Възможно кървене в коремните органи, стомашно-чревно кървене

ПРИЛАГАНА КОАГУЛОПАТИЯ
особеност - полидефицитен
етиология
1. Имунно инхибиране на прокоагуланти (резус конфликт)
2. Липса на вит. K-зависими фактори на кръвосъсирването (7, 10, 9, 2)
а) нарушен синтез в червата (дисбактериоза, диария)
б) нарушаване на абсорбцията на вит. К (дефицит на жлъчката)
в) тежко увреждане на черния дроб
3. Предозиране на хепарин

хиперкоакулационна
УВЕЛИЧЕН КАПАЦИТЕТ НА КРЪВНО ФОРМИРАНЕ В КОРАБИТЕ
тромбоза
СИНДРОМ НА ЛЕДА

DIC-СИНДРОМ (СИНДРОМ НА РАЗРЕШЕНА ИНТРАВАСКУЛЯРНА КРОВА КОРУЗАЦИЯ)
КЛАСИРАНЕ
* Според клиничния курс
1) остър (моментните форми се характеризират с тежък ход)
2) хронична
* По разпространение
1) локализиран
2) обобщени

етиология
Инфекции, септични състояния
шок (със септична - смъртност от 100%)
Хирургични интервенции, изгаряния
Всички терминални състояния, спиране на сърцето
Остра интраваскуларна хемолиза
Акушерска патология (20-25%)
Хемобластоза (с о. \u200b\u200bЛевкемия - 33-45%)
Деструктивни процеси в паренхимните органи
Алергични реакции

Етапи на DIC
1) Хиперкоагулация (образуване на множество кръвни съсиреци поради активиране на коагулационната система)
2) Коагулопатия на потреблението (изчерпване на коагулантната система, прекомерна употреба на тромбоцити за образуването на тромби)
3) Хипокоагулация (намаляване на активността на коагулантите, активиране на антикоагуланти, активиране на фибринолиза)
4) Приключване (възстановяване, усложнения, смърт)

Патогенеза на DIC
1) хипертромбинемия (тромбопластин навлиза в кръвта в големи количества от увредените тъкани и стимулира образуването на тромбин). При инфекции, активираните моноцитни макрофаги синтезират свои собствени коагуланти (f.7, f.10, f.9, f.2)

Патогенеза на DIC
2) Масивна тромбоцитна агрегация (причинява развитието на тромбоцитопения и потреблението и се усложнява от кръвоизливи)
3) Травма и хемолиза на еритроцитите (това води до много ADP, което увеличава адхезията и агрегацията на тромбоцитите)

Патогенеза на DIC
4) "Хуморална протеазна експлозия" (при активиране на прокоагуланти, антикоагуланти, фибринолитики, протеини на каликреин-кининовата система, комплементната система в кръвта натрупва много продукти от разпадането на протеини, които са много токсични и увреждат съдовете и тъканите)

Патогенеза на DIC
5) Изчерпване на фибрино-лизинговата система
(допринася за тромбоза)
6) Изчерпване на факторите на кръвосъсирването
(причинява развитието на кръвоизливи)

Клиника на DIC
1. Хемокоагулационен шок
причината
* нарушена микроциркулация (причинява развитието на тъканна хипоксия)
* натрупване на токсични продукти на протеолиза
проявления
* понижаване на кръвното налягане
* понижаване на централното венозно налягане
* кървене (провокира хеморагичен шок)

Клиника на DIC
2. Хемостатични нарушения
а) хиперкоагулация
Основната проява е тромбоза.
Ин витро кръвта се коагулира
б) hypocoagulation
Основната проява е кървене.
(в същото време системата на фибринолиза се изчерпва)

Клиника на DIC
3. тромбоцитопения
Възниква поради образуването на голям брой кръвни съсиреци в съдовете (потребление на тромбоцитопения)

Момичето, след като страда от възпалено гърло се появи петехиален обрив по кожата на крайниците и багажника. Обективно: брой на тромбоцитите 80 G / l, антитромбоцитни антитела. Алергичните реакции от кой тип (според Coombs и Jella) са в основата на това заболяване?
@ II тип (хуморална цитотоксична)
Тип I (анафилактичен)
Тип III (имунокомплекс)
V тип (стимулиращ)
Прилепването на тромбоцитите към колагена се нарушава и се наблюдава кървене от малки съдове. Нарушението на която връзка хемостаза може да бъде толерирана при пациент?
@Васкуларен тромбоцит
Коагулация и фаза
Коагулационна фаза III
фибринолиза
Коагулационна фаза II
Пациентът е диагностициран с тромбоцитопения. Какви клинични прояви са характерни за нарушения на тромбоцитната хемостаза?
@ Petechia, екхимоза (синини)
hemarthrosises
хематом
Намалено време на кървене
Повишено време за съсирване на кръвта
Пациентът по време на изследването намери тромбоцитопатия. Посочете коя промяна играе важна роля в патогенезата на тромбоцитопатията?
@ Производство на патологични тромбоцити от костния мозък
Намалена антикоагулантна активност
Хиперактивиране на тромбоцитопоезата
Повишена концентрация на прокоагуланти в кръвта
Инхибиране на фибринолизата.
Преди операцията е установено, че лицето е имало време на кървене, увеличено до 10 минути. Каква липса на формирани елементи в състава на кръвта може да причини такива промени?
@ Брой на тромбоцитите
Червени кръвни клетки
моноцити
лимфоцити
Броят на белите кръвни клетки
При пациенти продължителната употреба на аспирин причинява кръвоизливи. Обективна: тромбоцитопения с нарушена функционална активност на тромбоцитите. Тромбоцитопатията в този случай е причинена от инхибиране на активността:
Циклооксигеназа
Цитохромоксидаза
липоксигеназа
Супероксидна дисмутаза
Фосфолипаза А2.
Пациентът е диагностициран с понижаване на производството на васкуларен фактор на съдовия ендотелиум на von Willebrand. Какво е нарушение на съдова тромбоцитна хемостаза, наблюдавана в този случай?
@ Нарушаване адхезията на тромбоцитите
Нарушаване на тромбоцитната агрегация
хиперкоакулационна
Нарушаване на фибриновата полимеризация
Укрепване на фибринолизата
Дете с хеморагичен обрив, развило се след остри респираторни вирусни инфекции, е диагностицирано с хеморагичен васкулит (болест на Shenlein-Henoch). Алергичните реакции от кой тип (според Coombs и Jella) са в основата на това заболяване?
Тип III (имунокомплекс)
Тип I (анафилактичен)
Тип II (хуморална цитотоксична)
Тип IV (клетъчна цитотоксична)
V тип (стимулиращ)
Дете с хеморагичен обрив, развило се след остри респираторни вирусни инфекции, е диагностицирано с хеморагичен васкулит (болест на Shenlein-Henoch). Какво причинява нарушения на хемостазата при това заболяване?
@ Увреждане на съдовата стена
Наследствен дефект на съединителната тъкан на съдовата стена
Наследствена антикоагулантна недостатъчност
Инхибиране на фибринолиза
Наследствен прокоагулативен дефицит
Жена, която страда от жлъчнокаменна болест, има хеморагичен синдром, причинен от дефицит на витамин К. Кои от следните фактори не са подходящи за хиповитаминоза К?
@ Stuart - Prouer (f. X)
Факторът на Willebrand
Фибриностабилизиране (е. XIII)
Фибриноген (е. I)
Тромбофилия (ускоряване на съсирването на кръвта) е установена при пациента по време на изследването. Какво може да причини нарушение?
@ Дефицит на протеолитични ензимни инхибитори
Повишена концентрация на простациклин
Намалена концентрация на тромбин в кръвта
Повишени нива на хепарин в кръвта
Повишаване на концентрацията на фибринолизин в кръвта
Пациент с чернодробно заболяване намалява количеството на протромбин в кръвта. Това ще доведе до нарушение преди всичко:
@ Втората фаза на коагулационната хемостаза
фибринолиза
Третата фаза на коагулационната хемостаза
Съдова тромбоцитна хемостаза
Първата фаза на коагулационната хемостаза
Едно момче на 7 години, след падане от велосипед, се появи хемартроза на коляното. Въвеждането на криопреципитат и изпомпване на кръв от ставата е довело до значително подобряване на състоянието на детето. Какво заболяване трябва да мисля?
@Емофилия А
Хеморагичен васкулит.
thrombocytopathy
тромбоцитопения
Ревматоиден артрит
Момче с изразен хеморагичен синдром в кръвната плазма няма антигемофилия глобулин А (фактор VIII). Каква е основната фаза на хемостаза при това дете?
Вътрешен път на активиране на протромбиназа
Втягане на кръвен съсирек
Активиране на външния път на протромбиназа
Момчето страда от хемофилия. Какви са клиничните признаци на нарушение на коагулационната хемостаза?
Петехиален кръвоизлив
microhematuria
Ехимоза (синини)
Зрителна острота
@ Хематоми, продължително кървене
Жертва на инцидент някой път след претърпяване на политравма развил синдром на дисеминирана интраваскуларна коагулация (DIC). Какъв фактор беше инициаторът на това усложнение?
Тромбопластин в тъканите (е. III)
Фибриноген (е. I)
Антигемофилен глобулин А (е. VIII)
Пациент с остър панкреатит развива синдром на дисеминирана интраваскуларна коагулация (DIC). Каква същност е инициаторът на това усложнение?
@ Трипсин
Фибриноген (е. I)
Антигемофилен глобулин А (е. VIII)
Stuart Factor - Prouer (е. X)
Антигемофилен глобулин B (f. IX)
При пациенти с политравма и остра бъбречна недостатъчност, състоянието се усложнява от вътрешно кървене. Каква е основната връзка в патогенезата на този етап на DIC?
@ Консумация на фактори на кръвосъсирването
Освобождаването на левкоцити от депото
тромбоцитоза
Активиране на протромбиназа
Инхибиране на фибринолиза
Пациент с изгарящо заболяване е развил DIC синдром като усложнение. Кой етап от синдрома на DIC може да се подозира, ако е известно, че кръвта на пациента се коагулира за по-малко от 3 минути?
@ хиперкоагулация
gipokoagulyatsii
възстановяване
латентно
терминал
Пациент с нараняване на DIC - синдром. Какви промени в хемостазата се наблюдават във втората фаза на DIC синдрома?
@ Hypocoagulation
Хиперкоагулация.
фибринолиза
тромбоцитопения
thrombocytopathy
Пациент след преливане на несъвместима кръв имаше DIC - синдром. Каква е основната връзка в патогенезата на това усложнение при хемолизата?
Прием на вътреклетъчни протеази в кръвта
Натрупване на билирубин в кръвта
Прекомерна тромбопластинова кръв
Излишък на кръв протромбин
Увеличаването на съдържанието на плазминоген
Пациент с хронична бъбречна недостатъчност е развил DIC. При изследването се установи увеличение на времето за съсирване на кръвта, тромбоцитопения, повишени нива на фибрин-мономерни комплекси и продукти на разграждане на фибрина. На кой етап от DIC трябва да се мисли?
@ gipokoagulyatsii
хиперкоакулационна
възстановяване
латентно
нестабилен
Пациент с хронична лимфоцитна левкемия разви кръвоизливи в резултат на развитието на DIC синдром. Какви промени в периферната кръв ще се наблюдават?
@ Хипокоагулация, тромбоцитопения
Еритроцитоза, повишен вискозитет на кръвта
Тромбоцитоза, намалено време на кръвосъсирване
Хиперкоагулация, повишена тромбоцитна агрегация
Увеличава се прокоагулантната активност
Една жена със сепсис разви петехиални кръвоизливи, намален брой тромбоцити и фибриноген в кръвта и се появяват продукти на разграждане на фибрина. Какво може да бъде причината за отбелязаните промени?
@DIC синдром
левкопения
лимфопения
тромбоцитоза
 Повишена способност на кръвта да образува съсиреци в съдовете
Повишена способност на кръвта да образува съсиреци в съдовете Големи и малки букви на руски език
Големи и малки букви на руски език Skyrim Quest: Времеви трудности
Skyrim Quest: Времеви трудности