Какие бывают наследственные заболевания человека. Генетическое заболевание
ЭТО ПЕРЕПОСТ! Оригинал взят у eka_tyryshkina в Ошибки природы: люди с редкими заболеваниями.
Заболела я тут на днях, как всегда, как нам только куда надо идти в гости, я болею! Толи моя болезнь реагирует на плановость мероприятия, толи еще на что то, но хорошо что не реагирует на работу. Вообщем болезнь у меня не простая)
И вот болея дома в поздний час, уже переделав все дела, перечитав и пролистав все интресные сайты, вдруг не ожиданно для себя решила узнать о самых редких болязнях на планете и знаете, столько интересного и шокируешего!!!
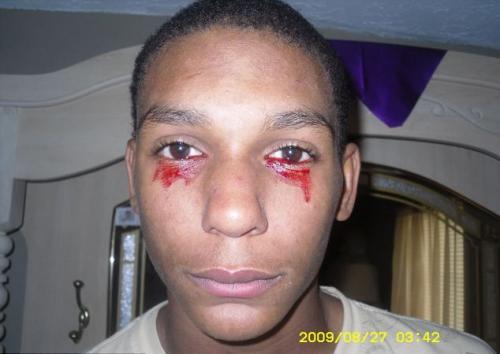
Гемолакрия
(«кровавые слезы») наблюдается у одного человека на миллион.Кровь, вместо слезной жидкости, начинает течь из глаз внезапно, и это может продолжаться около часа. За день больной обливается кровавыми слезами от 3 до 20 раз.
Точная причина этого заболевания до конца не изучена, а стало быть и лечению не поддаётся. Медицинские специалисты пока выдвигают версии что гемолакрия это одно из заболеваний крови или опухоли.
На фото - 15-летний Кальвино Инмэн (штат Теннеси, США)

Синдром Вампира
С диагнозом «синдром вампира»
(эктодермальная дисплазия) в мире насчитывается всего 7 тысяч человек.
Помимо мертвенно-бледной кожи и острых клыков (при отсутствии части зубов), у больных редкие и тонкие волосы, способность потеть снижена, поэтому их организм подвержен перегреву. Симптомы проявляются в детстве, однако выявить заболевание можно уже на стадии беременности с помощью генетических тестов.
Мальчики вынуждены носить темные очки и пользоваться солнцезащитным кремом, когда выходят на улицу, поскольку они не могут находиться под прямыми солнечными лучами.
При этом физическое развитие и двигательная активность остается в норме.
Сама болезнь неизлечима, коррекции поддаются только симптомы. В частности, можно восстановить нормальную форму зубов.
Болезнь Саймона была диагностирована в младенчестве. Когда Мэнди была беременна второй раз, ее предупредили, что у второго ребенка может быть такое же заболевание.
Однако Саймон рос и развивался хорошо, поэтому родители пошли на этот риск.
Мальчики говорят: "некоторые дети смеются над нашей внешностью, но наши друзья думают, что это круто"
На фото - Саймон (13 лет) и Джордж (11 лет) Каллен (Саффолк, Великобритания).

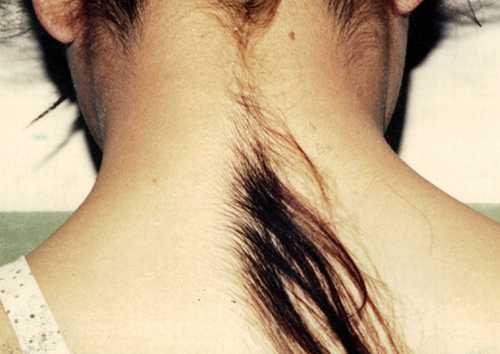
Гипертрихоз
(«синдром оборотня») - это заболевание, проявляющееся в избыточном росте волос, не свойственному данному участку кожи, не соответствующему полу и возрасту.Во всем мире зарегистрировано всего чуть более сорока таких пациентов, так что самый удобный для них способ заработать - демонстрировать свое уродство...Они подают заявки в книгу рекордов Гиннеса,- чтобы прославится и заработать деньги... Китайцу Юй Чжэньхуан это удалось на все сто - благодаря своей сверхволосатости он основал популярнейшую в своей стране рок-группу и стал миллионером.
Неизвестно, отчего происходит подобная мутация. И лечение от гипертрихоза тоже пока еще никто не разработал. Косметологи умеют только удалять волосы на достаточно долгий срок...
На фото - 6-летняя Нат Сасуфан
(Таиланд), 2007 год

На фото - 33-летний Юй Чжэньхуан (Китай), самый волосатый человек в мире

Слоновая болезнь
(«синдром Протея», слоновость, элефантиаз, элефантиазис) - увеличение размеров какой-либо части тела за счёт болезненного разрастания кожи и подкожной клетчатки.Всего в мире насчитывается примерно 120 человек с этим неизлечимым заболеванием…
А самым известным больным был «человек-слон» - Джозеф Меррик. О знаменитом британце в 1980 году режиссёр Дэвид Линч даже снял фильм, который был выдвинут на «Оскар» по восьми номинациям… Речь в фильме шла о человеческом достоинстве… Грим Джона Херта, который и сыграл Меррика, был создан на основе представленного в Королевской больнице Лондона заспиртованного тела Джозефа Меррика. Его накладка ежедневно занимала у актёра 12 часов в день…
На фото - 35-летняя Менди Селларс
(Великобритания)

Генную аномалию, заключающуюся в ускоренном старении организма
,
- прогерию
- подразделяют на детскую (синдром Гетчинсона) и взрослую (синдром Вернера). Впервые о синдроме преждевременного старения заговорили 100 лет назад. И не удивительно, такие случаи встречаются один раз на 4-8 миллионов младенцев. Прогерия (от греческого prо - раньше, gerontos - старец) - крайне редкое генетическое заболевание, ускоряющее процесс старения примерно в 8-10 раз. Проще говоря, ребенок за один год стареет на 10-15 лет. Восьмилетний выглядит на 80 лет - с сухой морщинистой кожей, облысевшей головой... Эти дети обычно погибают в 13-14 лет после нескольких инфарктов и инсультов на фоне прогрессирующего атеросклероза, катаракты, глаукомы, полной потери зубов и т.д. И лишь немногие живут до 20 лет или дольше.
Сейчас в мире известны всего 42 случая заболевания людей прогерией... Из них 14 человек проживают на территории Соединенных Штатов, 5 - в Росии, остальные в Европе...
В настоящее время существует несколько организаций, оказывающих помощь маленьким старичкам и их семьям. В Интернете есть сайты, посвященные именно этой проблеме, некоторые из них открыты медиками или социальными работниками, другие - семьями больных.
На фото - 24-летний Леон Бот



38-летний человек-дерево
Деде Косвара, проживающий на острове Ява, в Индонезии, стал знаменитым на весь мир из-за вируса папилломы человека, который обычно приводит к появлению небольших бородавок, но в случае с индонезйцем до неузнаваемости деформировал его конечности.
Проблема Дедэ состояла в том, что он имел редкое генетическое отклонение, которое не позволяло его иммунной системе сдержать рост этих бородавок. Поэтому вирус смог "завладеть клеточным механизмом его клеток кожи", отдавая им приказы вырабатывать большое количество рогового вещества, из которого и состояли. У Дедэ обнаружилось также низкое содержание лейкоцитов в крови.


Болезнь Бабочки
Буллёзный эпидермолиз в гиперпластической форме — это генетическое заболевание, которое проявляется в первые дни жизни. По сути, кожа новорождённого настолько нежная, что любое прикосновение приводит к возникновению ран и пузырей. Больше всего страдают выступающие участки: локти, колени, стопы, руки. Возникшая язва, с которой слоями сходит кожа, долго не заживает, из неё выделяется жидкость. После образуется большой малиновый шрам.
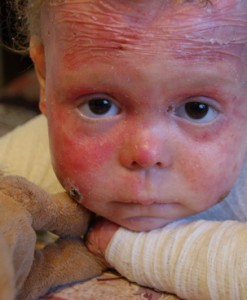
Лечения данного заболевания нет, возможно только облегчение симптомов. Не так давно на всю Россию прогремела история Лизы Кунигель, которая живёт с буллёзным эпидермолизом уже почти десять лет. Несколько раз в день ей необходимы перевязки и обработка противомикробными мазями и гелями. Кроме этого, все 9 лет Лизу сопровождает боль.
Синдром русалки
Одной из редчайших аномалий в развитии является сиреномелия, в народе называемая «синдромом русалки». При данном дефекте новорожденные появляются на свет со срощенными ногами, похожими на рыбий хвост. У них функционирует только одна почка, отсутствуют гениталии. Из-за обширного поражения внутренних органов такие младенцы обычно вскоре умирают. Болезнь встречается у одного из 100 000 новорожденных. За все годы наблюдений лишь трое малышей смогли выжить. Одной из них была Шайло Пепин.
Шайло родилась в 1999 году и стала самым знаменитым ребенком с «синдромом русалки». За те 10 лет, что она смогла прожить у нее появились тысячи друзей по всему миру, которые поддерживали девочку и её маму. Шайло старалась вести полноценную жизнь - она, как и все обычные дети ходила в школу, посещала занятия танцев, ездила в парки развлечений. Известной девочка стала после участия в шоу Опры Уинфри. Learning Chanel снял о ней несколько фильмов, ей посвящены сотни сайтов в интернете.

История с Шайло - удивительная история о чуде. Ребенок, который все свое детство боролся за то, что выжить. Маленькая девочка, умевшая радоваться каждому дню, несмотря на неизлечимую болезнь.
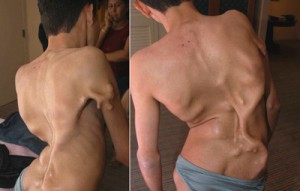
Болезнь Мюнхеймера
Фибродисплазия - заболевание крайне редкое. Официальная статистика такова: 1 больной на 2 000 000 человек. Болезнь Мюнхеймера возникает в результате мутации гена и при рождении проявляется во внешних дефектах. У младенца искривлены большие пальцы стоп, позвоночник. Патология приводит к инвалидности, ранней смертности. Там где должны проходить противовоспалительные процессы, начинает образовываться костный нарост, поэтому часто заболевание называют «болезнью второго скелета».
Любой, даже незначительный ушиб, может привести к развитию остекленения на пораженном месте.На сегодняшний момент официального лечения от смертельной болезни не существует. Ученые разработали препарат, который теоретически может бороться с недугом. Однако, необходимых клинических исследований проведено ещё не было. Увы, провести их очень трудно - во всем мире насчитывается не более 600 человек с болезнью Мюнхеймера.
Феномен «Линий Блашко» характеризуется наличием странных полос по всему телу. Линии Блашко - это невидимый рисунок, заложенный в ДНК. И проявлением заболевания становится видимость этого рисунка.
Обычно рисунок на спине имеет V-образную форму, а на груди, животе и на боках - S-образную.
Причиной заболевания может являться мозаицизм. В любом случае, появление линий Блашко никак не связано с нервной, мускульной и лимфатической системами человека.
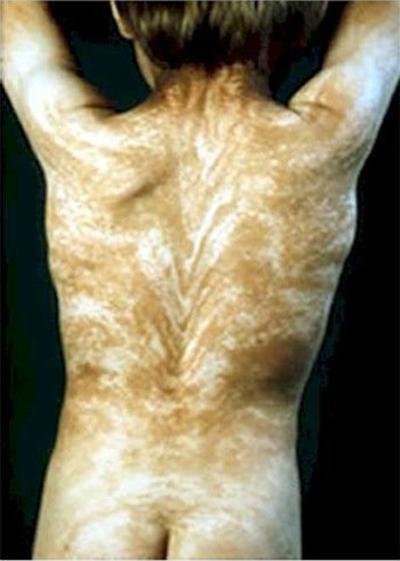
Еще одно аномальное заболевание - акантокератодермия , или «синдром синей кожи». Люди с таким диагнозом могут иметь голубую, цвета индиго, сливовую или практически фиолетовую кожу.В 60-х годах прошлого века в штате Кентукки проживало целое семейство «синих» людей. Они были известны как Синие Фьюгейты. Эта особенность передавалась из поколения в поколение.


Редкими заболеваниями страдает около 6% жителей Земли, и это число продолжает увеличиваться. Все уникальные болезни имеют различную природу, однако подавляющее большинство феноменальных недугов связано с генетическими аномалиями и инфекциями.
По наследству могут передаваться не только внешние признаки, но и заболевания. Сбои в генах предков приводят, в итоге, к последствиям в потомстве. Мы расскажем о семи самых распространенных генетических заболеваниях.
Наследственные свойства передаются потомкам от предков в виде генов, объединенных в блоки, которые называются хромосомами. Все клетки организма, за исключением половых, имеют двойной набор хромосом, половина которого достается от матери, а вторая часть – от отца. Болезни, чей причиной служат те или иные сбои в генах, являются наследственными.
Близорукость
Или миопия. Генетически обусловленное заболевание, суть которого состоит в том, что изображение формируется не на сетчатке глаза, а перед ней. Самой распространенной причиной такого явления считается увеличенное в длину глазное яблоко. Как правило, близорукость развивается в подростковом возрасте. Человек при этом прекрасно видит вблизи, но плохо видит вдаль.
Если оба родителя близоруки, то риск развития миопии у их детей составляет свыше 50% . Если оба родителя обладают нормальным зрением, то вероятность развития близорукости составляет, не более 10%.
Исследуя близорукость, сотрудники Австралийского национального университета в Канберре пришли к выводу, что миопия присуща 30% представителей европеоидной расы и поражает до 80% уроженцев Азии, включая жителей Китая, Японии, Южной Кореи и т. д. Собрав данные более 45 тыс. человек, ученые идентифицировали 24 гена, связанных с близорукостью, а также подтвердили их связь с двумя ранее установленными генами. Все эти гены отвечают за развитие глаза, его структуру, передачу сигналов в ткани глаза.
Синдром Дауна
Синдром, получивший свое название в честь английского врача Джона Дауна, который впервые описал его в 1866 году, представляет собой одну из форм хромосомной мутации. Синдрому Дауна подвержены все расы.
Болезнь является следствием того, что в клетках присутствуют не два, а три экземпляра 21-й хромосомы. Генетики называют это трисомией. В большинстве случаев лишняя хромосома передается ребенку от матери. Принято считать, что риск рождения ребенка с синдромом Дауна зависит от возраста матери. Однако из-за того, что в целом чаще всего рожают в молодости, 80% всех детей с синдромом Дауна рождены женщинами в возрасте до 30 лет.
В отличие от генных, хромосомные нарушения являются случайным сбоем. И в семье может быть лишь один человек, страдающий подобным заболеванием. Но и здесь случаются исключения: в 3-5% случаев наблюдаются более редкие – транслокационные формы синдрома Дауна, когда у ребенка имеется более сложная структура набора хромосом. Подобный вариант болезни может повторяться в нескольких поколениях одной семьи.
Согласно информации благотворительного фонда «Даунсайд Ап», в России ежегодно рождается порядка 2500 детей с синдромом Дауна.
Синдром Клайнфельтера
Еще одно хромосомное нарушение. Примерно на каждые 500 новорожденных мальчиков приходится один с этой патологией. Синдром Клайнфельтера проявляется, как правило, после полового созревания. Страдающие этим синдромом мужчины бесплодны. Кроме того, им присуща гинекомастия – увеличение грудной железы с гипертрофией желез и жировой ткани.
Синдром получил свое название в честь американского врача Гарри Клайнфельтера, впервые описавшего клиническую картину патологии в 1942 году. Совместно с эндокринологом Фуллером Олбрайтом он выяснил, что если в норме женщины имеют пару половых хромосом ХХ, а мужчины – ХУ, то при данном синдроме у мужчин присутствует от одной до трех дополнительных Х-хромосом.
Дальтонизм
Или цветовая слепота. Носит наследственный, гораздо реже приобретенный характер. Выражается в неспособности различать один либо несколько цветов.
Дальтонизм связан с X-хромосомой и передается от матери, обладательницы «поломанного» гена к сыну. Соответственно, дальтонизмом страдают до 8% мужчин и не более 0,4% женщин. Дело в том, что у мужчин «брак» в единственной X-хромосоме не компенсируется, поскольку второй X-хромосомы, в отличие от женщин, у них нет.
Гемофилия
Еще одна болезнь, наследуемая сыновьями от матерей. Широко известна история потомков английской королевы Виктории из династии Виндзоров. Ни она сама, ни ее родители не страдали этим тяжелым заболеванием, связанным с нарушением свертывания крови. Предположительно, мутация гена произошла спонтанно, из-за того, что отцу Виктории на момент ее зачатия было уже 52 года.
От Виктории «роковой» ген унаследовали дети. Ее сын Леопольд умер из-за гемофилии в 30 лет, а две из ее пяти дочерей, Алиса и Беатриса, были носителями злосчастного гена. Одним из самых известных потомков Виктории, страдавших гемофилией, является сын ее внучки, царевич Алексей, единственный сын последнего российского императора Николая II.
Муковисцидоз
Наследственное заболевание, которое проявляется в нарушении работы желез внешней секреции. Оно характеризуется повышенным потоотделением, выделением слизи, которая накапливается в организме и мешает ребенку развиваться, а, главное, препятствует полноценной работе легких. Вероятен летальный исход из-за дыхательной недостаточности.
Согласно данным российского филиала американской химико- фармацевтической корпорации Abbott, средняя продолжительность жизни больных муковисцидозом составляет в европейских странах 40 лет, в Канаде и США – 48 лет, в России – 30 лет. Из известных примеров стоит привести французского певца Грегори Лемаршаля, умершего в 23 года. Предположительно, муковисцидозом страдал и Фредерик Шопен, скончавшийся в результате отказа легких в возрасте 39 лет.
Заболевание, упоминаемое в древнеегипетских папирусах. Характерный симптом мигрени – эпизодические или регулярные сильные приступы головной боли в одной половине головы. Римский врач греческого происхождения Гален, живший во II веке, назвал болезнь гемикранией, что переводится как «половина головы». От этого термина и произошло слово «мигрень». В 90-х гг. ХХ века было установлено, что мигрень преимущественно обусловлена генетическими факторами. Был открыт ряд генов, ответственных за передачу мигрени по наследству.
К ребенку от его биологических родителей могут передаваться не только внешние черты и особенности характера, но и ряд проблем со здоровьем.
Наследственные заболевания встречаются редко, но, как правило, это довольно тяжелые болезни, которые практически не поддаются лечению.
Каждый ген организма человека содержит в себе уникальную ДНК, он имеет свой уникальный код конкретного признака.
В данной ситуации необходимо обратиться за помощью к врачу – генетику, пройти генетическую консультацию, чтобы узнать степень риска возникновения того или иного генетического заболевания
Болезнь Дауна
На сегодняшний день одной из наиболее распространенных болезней, которая

передается по наследству, является Болезнь Дауна. Статистические данные показывают, что такое заболевание встречается у одного новорожденного из семисот малышей. Данный диагноз, как правило, устанавливается специалистом еще в родильном доме, на сроке 3-5 дней жизни новорожденного ребенка.
Для того, чтобы подтвердить данный диагноз, проводится такая процедура, как исследование кариотипа. Она заключается в исследовании набора хромосом у новорожденного ребенка. Ребенок, который болеет , имеет семь хромосом, что на одну больше, чем у здорового человека. Такое заболевание встречается, как у мальчиков, так и у девочек, пол в данном случае не играет никакой роли.
Болезнь Шершевского-Тернера
Данное заболевание характерно только для детей женского пола. Первые признаки данной генетической патологии можно обнаружить в возрасте 10-12 лет.
Как правило, на затылке волосы растут очень медленно, причем, они имеют глубоко – посаженный корень. В возрасте 15 – 16 лет, а то и старше, у девочек отсутвуют , именно это и является причиной обращения к специалисту. С возрастом, заболевание может стать причиной проявления некоторых проблем с умственным развитием ребенка. Генетическая структура болезни Шершевского-Тернера у девочек характеризуется отсутствием одной Х хромосомы.
Болезнь Клайнфельтера
Болезнь Клайнфельтера — это генетическая патология, которая проявляется только у мальчиков. Первые признаки заболевания можно будет обнаружить при достижении ребенком 15 – 16 – летнего возраста.
Первые признаки:

При исследовании на хромосомы болезнь Клайнфельтера характеризуется их увеличенным количеством: одной хромосомой Х больше. В некоторых случаях, могут дополнительно присутствовать и другие хромосомы: У, ХХ, ХУ.
Многофакторные генетические заболевания
Многофакторные генетические заболевания представляют собой генетические патологии, которые могут проявиться у новорожденного ребенка в любой семье.
В данном случае причиной развития таких заболеваний выступают не только генетические отклонения, но и ряд посторонних факторов, например, плохая экология, нарушенный ритм жизни родителей.
К числу таких заболеваний относится: ишемическая болезнь сердца, заболевания желудка, а также проблемы с кровеносной системой.
Врожденные дефекты, которые относятся к многофакторным генетическим заболеваниям — это заячья губа, волчья пасть и расщепление позвоночника.
В настоящее время все многие патологии можно выявить с помощью современного оборудования: ультразвуковое исследование плода сможет показать многие аномалии в развитии ребенка.
Генетические заболевания – это редкие и сложные по своей природе болезни, которые практически не поддаются лечению из-за нарушений на геном уровне. Поэтому специалисты рекомендуют при планировании ребенка заранее проходить консультацию с генетиком во избежание проблем в будущем.
О генетических болезнях расскажут специалисты на видео:
Заметили ошибку? Выделите ее и нажмите Ctrl+Enter , чтобы сообщить нам.
Понравилось? Лайкни и сохрани у себя на страничке!
Генетические заболевания уникальны тем, что не зависят от образа жизни человека, от них нельзя застраховаться просто перестав есть жирную пищу или начав делать зарядку по утрам. Они возникают в результате мутации и могут передаваться из поколения в поколение.
Редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в 40 семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего - после 50 лет) и продолжается от 7 до 36 месяцев. По мере того, как заболевание прогрессирует, пациент страдает от всё более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает - как правило, от истощения или пневмонии.

Синдром нарколепсии-катаплексии, для которого характерны внезапные приступы сна и расслабления мускулатуры тела, тоже имеет генетическую природу и возникает из–за нарушений быстрой фазы сна. Он встречается намного чаще фатальной семейной бессонницы: у 40 из каждых 100 тыс. человек, в равной степени у мужчин и у женщин. Человек, страдающий нарколепсией, способен внезапно заснуть на несколько минут посреди дня. «Сонные атаки» напоминают фазу быстрого сна и могут случаться очень часто: до 100 раз в день, с предшествующей им головной болью, либо без неё. Они часто провоцируются бездеятельностью, но могут возникать в совершенно неподходящее время: во время полового акта, занятий спортом, вождения. Просыпается человек отдохнувшим.
![]()
Синдром Юнера Тана (СЮТ) характерен прежде всего тем, что люди, страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего люди с СЮТ пользуются примитивной речью и имеют врождённую мозговую недостаточность. В 2006-м году о семье Улас был снят документальный фильм под названием «Семья, ходящая на четвереньках». Тан описывает это так: «Генетическая природа синдрома предполагает обратную ступень в эволюции человека, вызванную, скорее всего, генетической мутацией, обратному процессу перехода от квадропедализма (хождения на четырёх конечностях) к бипедализму (хождению на двух). В этом случае синдром соответствует теории прерывистого равновесия.

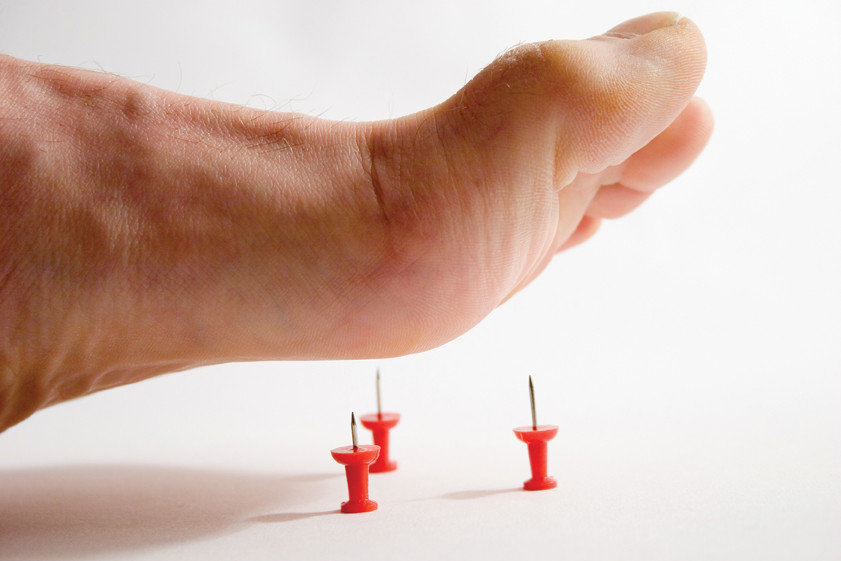
Одно из самых редких в мире заболеваний: этот вид нейропатии диагностируют у двух человек из миллиона. Аномалия возникает из-за поражения периферической нервной системы, возникающего вследствие переизбытка гена PMP22. Главным признаком развития наследственной сенсорной нейропатии первого типа является потеря чувствительности рук и ног. Человек перестаёт испытывать боль и ощущать изменение температуры, что может привести к возникновению некроза тканей, например, если вовремя не распознать перелом или другую травму. Боль - одна из реакций организма, сигнализирующих о каких-либо «неполадках», поэтому потеря болевой чувствительности чревата слишком поздним выявлением опасных заболеваний, будь то инфекции или язвы.
Люди, страдающие этим необычным недугом, выглядят значительно старше своего возраста, поэтому его иногда называют «обратный синдром Бенджамина Баттона». Из-за наследственной генетической мутации, а иногда в результате применения некоторых лекарственных препаратов в организме нарушаются аутоиммунные механизмы, что приводит к быстрой потере подкожных жировых запасов. Чаще всего страдает жировая ткань лица, шеи, верхних конечностей и туловища, вследствие чего возникают морщины и складки. Пока подтверждено лишь 200 случаев прогрессирующей липодистрофии, и главным образом она развивается у женщин. При лечении врачи используют инсулин, «подтяжки» лица и инъекции коллагена, однако это даёт лишь временный эффект.

Гипертрихоз также называют «синдромом оборотня» или «синдромом Абрамса». Он проявляется только у одного человека из миллиарда, и только 50 случаев со времён Средневековья были задокументированы. Люди, страдающие гипертрихозом, отличаются чрезмерным количеством волос на лице, ушах и плечах. Это происходит из-за нарушения связей между эпидермисом и дермой во время формирования у трёхмесячного плода волосяных фолликул. Как правило, сигналы от образующейся дермы «сообщают» фолликулам их форму. Фолликулы тоже, в свою очередь, сигнализируют кожным слоям, что в этой области одна фолликула уже есть, и это приводит к тому, что на теле волоски растут на приблизительно одинаковом расстоянии друг от друга. В случае с гипертрихозом эти связи нарушены, что приводит к образованию слишком плотного волосяного покрова на тех участках тела, где его быть не должно.

Если вы когда-нибудь слышали о козьем обмороке, то примерно знаете, как выглядит конгенитальная миотония - из-за мышечных спазмов человек на некоторое время будто замирает. Причиной возникновения конгенитальной (врождённой) миотонии является генетическое отклонение: вследствие мутации нарушается работа хлорных каналов скелетных мышц. Мышечная ткань оказывается «сбитой с толку», возникают произвольные сокращения и расслабления, причём патология может затрагивать мускулатуру ног, рук, челюстей и диафрагмы.

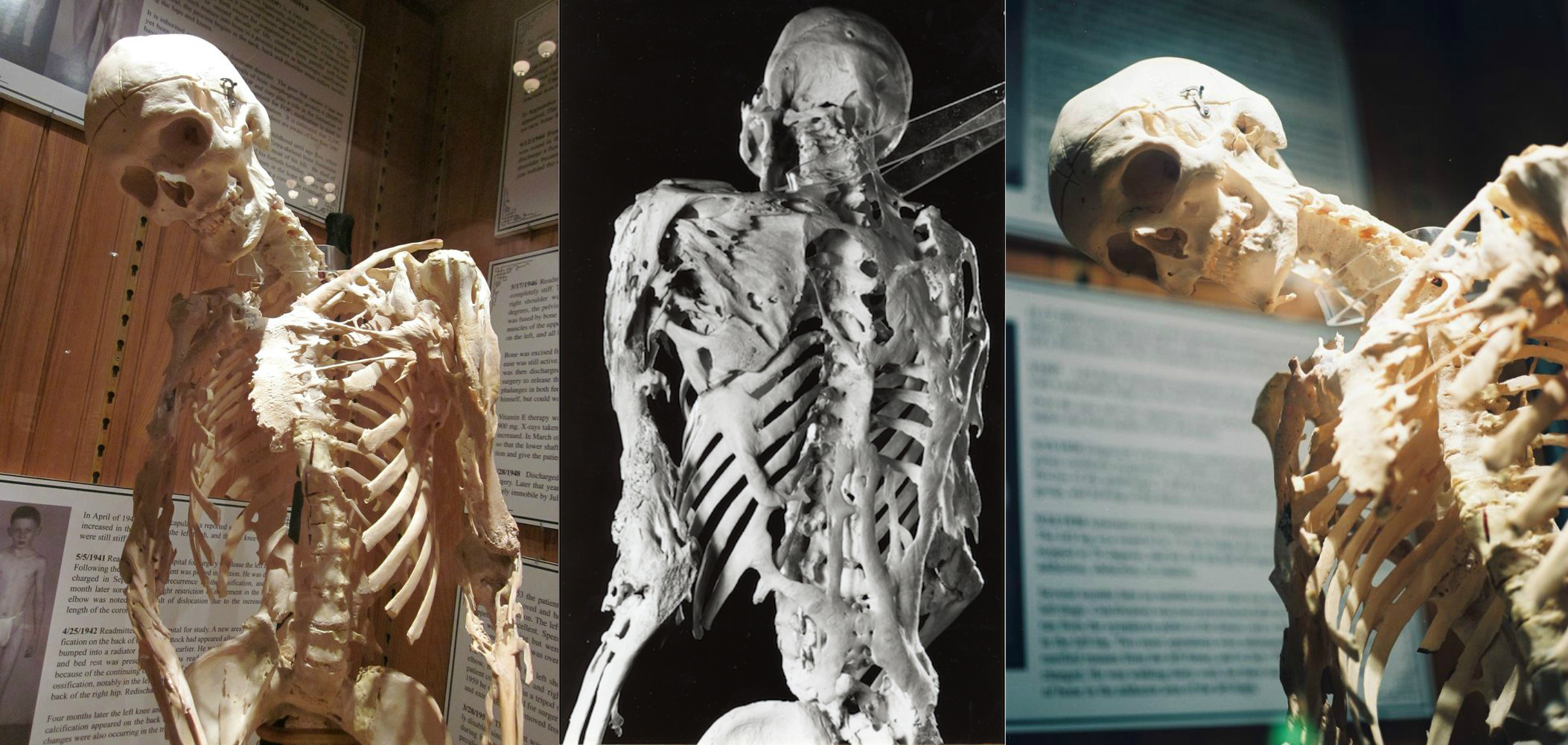
Редкое генетическое заболевание, при котором организм начинает формировать новые кости - оссификаты - в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из–за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из–за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.
Это наследственное заболевание кожи проявляется в повышенной чувствительности человека к ультрафиолетовым лучам. Возникает оно из-за мутации белков, ответственных за исправление повреждений ДНК, появляющихся при воздействии ультрафиолетового излучения. Первые симптомы обычно проявляются в раннем детстве (до 3-х лет): когда ребёнок находится на солнце, у него возникают серьёзные ожоги уже после нескольких минут воздействия солнечных лучей. Также заболевание характеризуется появлением веснушек, сухостью кожи и неравномерным изменением цвета кожного покрова. Согласно статистике, люди с пигментной ксеродермой более других подвержены риску развития онкологических заболеваний: при отсутствии надлежащих профилактических мер, примерно у половины детей, страдающих ксеродермой, к десяти годам развиваются те или иные раковые заболевания. Существует восемь видов этого недуга различной тяжести и симптомов. По данным европейских и американских медиков, болезнь встречается примерно у четырёх человек из миллиона.

Любопытное название для болезни, не так ли? Впрочем, существует и научный термин для обозначения этой «болячки» - десквамативный глоссит. Географический язык проявляется примерно у 2,58% людей, причём чаще всего заболевание имеет хронические свойства и обостряется после еды, во время стресса или гормональных стрессов. Симптомы проявляются в возникновении на языке обесцвеченных гладких пятен, напоминающих острова, потому заболевание и получило столь необыкновенное прозвище, причём со временем некоторые «острова» меняют свою форму и расположение, в зависимости от того, какие из вкусовых сосочков, расположенных на языке, заживают, а какие, наоборот, раздражаются.
Географический язык практически безвреден, если не брать в расчёт повышенную чувствительность к острой пище или некоторый дискомфорт, который он может причинять. Медицине неизвестны причины возникновения этой болезни, но есть данные о генетической предрасположенности к её развитию.
Будучи довольно редким недугом, встречающимся 1 раз на 2 миллиона, синдром каменного человека, известный в медицине как прогрессирующая оссифицирующая фибродисплазия (ФОП), является генетическим заболеванием, вызванным мутацией в генах, которая позволяет травмированной соединительной ткани превращаться в кость. У людей, страдающих каменной болезнью, создаётся новая структура скелета. Перерождение начинается с костей шеи и распространяется вниз, затрагивая все структуры, включая крестцовую кость.
Ранее были зарегистрированы несколько случаев этого заболевания. Скелеты, на которых видны окостенения соединительной ткани, можно найти в музее медицинской истории города Мюттера, расположенного в Филадельфии, США и в Лондоне. На самом деле они являются частью коллекции музея «Хантериан» при Королевском Колледже Хирургов.
Синдром каменного человека начинает проявляться в раннем возрасте, с годами развиваются все новые и новые кости. Большинству пациентов удаётся прожить около 40 лет, так как после этого возраста вырастает вероятность смертельного исхода в результате проблем с дыханием. Спортивные травмы, травмы от падения или даже инвазивные медицинские процедуры опасны для таких людей, так как могут спровоцировать интенсивное воспаление мышц, сухожилий и связок, вместо которых, несмотря на исцеление, образуются кости, заменяющие травмированные соединительные ткани.
Причины возникновения
Синдром каменного человека — аутосомно-доминантное генетическое заболевание, вызванное мутацией в гене ACVR 1(активин-рецептор типа 1). В большинстве случаев это расстройство носит спонтанный характер, и новые мутации возникают при отсутствии семейного анамнеза заболевания. Они могут появляться в результате воздействия ионизирующей радиации, химических веществ, лекарств и инфекций. Это воздействие может произойти в утробе матери или вскоре после рождения. Однако в истории болезни были зафиксированы несколько эпизодов передачи гена от родителя к потомку. При таких обстоятельствах для проявления болезни достаточно одного изменённого аллеля (копии) родительского гена.
В обычных условиях ген ACVR 1 действует как модулятор, который контролирует рост и размножение клеток мышц, связок и других соединительных тканей. Он кодирует костный морфогенетический белок ВМП 1, управляющий нормальной оссификацией и созреванием костей скелета. При мутации вследствие травм или вирусной инфекции в мышцах и соединительных тканях происходит постоянная активация гена с последующим выпуском неполноценных белков. Это приводит к осаждению клеток костной ткани в повреждённых местах, избыточному росту кости и сращению суставов и костей.
Симптомы
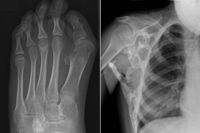
Симптомы синдрома каменного человека достаточно отчётливы и развиваются на протяжении всей жизни. К ним относятся:
- Наличие искажённых больших пальцев ног считается первым признаком данного расстройства. Это также важный симптом, так как он помогает врачам дифференцировать его от других аналогичных состояний опорно-двигательного аппарата.
- Затруднение в движении и тугоподвижность суставов, возникшая в результате их слияния с новообразовавшимися костями.
- Сложности с приёмом пищи по причине слияния челюстных костей. Вследствие нарушения питания, как правило, возникают связанные с этим заболевания. Сращение челюстного сустава также приводит к проблемам с речью.
- С образованием новых костей в области грудной клетки, её расширение существенно усложняется, что приводит к проблемам с дыханием. Большинство пациентов с синдромом каменного человека умирают от дыхательной недостаточности и пневмонии.
- Люди, страдающие от данного заболевания, имеют характерный горб на спине, отклонение позвоночника в сторону, затруднение в движении и неспособность выполнять те или иные задачи. Тело начинает выглядеть как сплошная статуя. Чаще всего на 2-м или 3-м десятилетии жизни пациенты полностью становятся прикованными к постели.
Как с этим справляться
На сегодняшний день лечения синдрома каменного человека не существует. Впрочем, применение высоких доз кортикостероидов может помочь уменьшить интенсивность воспаления мышечной и соединительной ткани, тем самым задерживая формирование новых костей. Пациентам, как правило, рекомендуется избегать падений, травм и занятий контактным видом спорта.
Перинатальный тест для детей, рождённых с этим пороком, не является рутинной процедурой. Тем не менее, сам диагноз может быть подтверждён с помощью генетических исследований.
Синдром каменного человека у девочки-подростка

В течение многих лет Сини Немок, милая 18-летняя девушка из Северного Кенсингтона (Лондон) борется с редким генетическим заболеванием. Впервые диагноз «синдром каменного человека» был поставлен Сини в возрасте 12 лет. В результате небольшого падения воспалённый участок на спине постепенно заменялся костными новообразованиями и причинял мучительную боль. Шея и позвоночник слились в одно целое, не давая девушке возможности поднять руки выше талии. Её сегодняшняя жизнь омрачена постоянными страхами от падений и травм, так как это наверняка приведёт к прорастанию новых костей и ухудшит нынешнее состояние.
Несмотря на столь сложный диагноз, Сини по-прежнему проводит время со своими друзьями. Она любит готовить, делать покупки и как любая нормальная девочка-подросток, обожает макияж. Её семья не оставляет надежды на то что учёным всё-таки удастся понять этот сложный патологический процесс и создать лекарство для лечения синдрома каменного человека.
Сорокина Юлия Сергеевна
Как проявляется атопический дерматит в детском возрасте? У большинства детей первые признаки болезни возникают в грудном возрасте. Их появление связывают, как правило, с введением в питание искусственных смесей, коровьего молока, яиц, рыбы, некоторых круп. На лице, туловище, руках и ногах малышей появляются краснота, пузырьки, кожа мокнет, либо, наоборот, сохнет и шелушится. Дети становятся беспокойными, плохо спят. Если не упустить момент и прибегнуть к помощи дерматолога, аллергическое воспаление кожи можно остановить. Возраст до 3 лет является самым благодарным временем для лечения. В этом периоде детства можно с максимальной вероятностью добиться
У большинства детей первые признаки болезни возникают в грудном возрасте. Их появление связывают, как правило, с введением в питание искусственных смесей, коровьего молока, яиц, рыбы, некоторых круп. На лице, туловище, руках и ногах малышей появляются краснота, пузырьки, кожа мокнет, либо, наоборот, сохнет и шелушится. Дети становятся беспокойными, плохо спят. Если не упустить момент и прибегнуть к помощи дерматолога, аллергическое воспаление кожи можно остановить. Возраст до 3 лет является самым благодарным временем для лечения. В этом периоде детства можно с максимальной вероятностью добиться
прерывания «марша» атопии.
В возрасте 6 — 7 и 12 — 14 лет возможно обострение кожного процесса. Провоцирует его домашняя пыль, шерсть домашних животных, пыльца растений, бактерии, плесень, а пищевые аллергены отступают на второй план. Большую роль играют стрессовые ситуации, нарушение режима дня, перегрузка ребенка учебой, занятиями в многочисленных кружках. Воспаление переходит на сгибы рук, ног, шеи. Кожа становится более сухой, утолщается, в месте расчесов образуются корочки.
У некоторых людей проявления атопии в младенческом возрасте незначительны и остаются без внимания родителей и педиатров, но в зрелом возрасте возникают вновь, казалось бы «впервые». Воспаление кожи возникает чаще в сгибах конечностей, на пальцах рук, в области голеностопных суставов, на лице и шее. Высыпания могут быть мокнущими, по типу экземы или сухими. Объединяет их сильный зуд и остаточные явления в виде утолщенной грубой кожи, шелушения, сухости.
Атопический маршКак правило, развиваются позже проявлений атопического дерматита. В последние годы говорят о так называемом «атопическом марше». Что это такое? Атопический марш означает то, что «диатез» у детей может служить начальной стадией для развития других, более тяжелых форм аллергии — бронхиальной астмы, поллиноза (аллергия на пыльцу растений), пищевой и лекарственной аллергии, аллергического ринита (насморк) и конъюнктивита (воспаление глаз), болей в суставах, мигреней. Причем эти заболевания могут меняться в разные периоды жизни у одного человека. Например, были высыпания на коже, затем все прошло, и появился поллиноз или мигрень. Контролируя течение атопического дерматита, врачи и ученые рассчитывают предотвратить «атопический марш».
Атопия в наследство?Атопический дерматит и атопия — это врожденные и наследуемые состояния. Это значит, что существуют генетические факторы, определяющие атопию. Эти механизмы сегодня тщательно изучаются, чтобы послужить разработке новых методов лечения атопии в будущем.
Все пути наследования атопического дерматита пока не установлены окончательно. Мы знаем, что если у одного из родителей или ближайших родственников ребенка есть атопический дерматит, астма или аллергический ринит, то у самого ребенка атопический дерматит разовьется с вероятностью 50%. В то же время, у родственников 30% лиц с атопическим дерматитом не отмечается каких-либо проявлений аллергии.
Атопическая наследственность — вовсе не приговор, обрекающий человека всю жизнь или какое-то время испытывать симптомы аллергии. Можно быть «атопиком» и не иметь сыпи и зуда.
Внешний вид человека с атопиейКожа страдающих атопическим дерматитом отличается сухостью. Кожа туловища и разгибательных поверхностей рук и/или ног покрыта блестящими, телесного цвета мелкими «пупырышками». На боковых поверхностях плеч, локтях, иногда в области плечевых суставов могут быть роговые плотные мелкие папулы («тёрка»). В старшем возрасте кожа отличается пестротой с наличием темных и белых пятен. Нередко у больных в области щек определяются белесоватые пятна.
В период ремиссии единственными минимальными проявлениями атопического дерматита могут быть едва шелушащаяся слегка утолщенная кожа или даже трещины в области прикрепления ушной раковины. Кроме того, таковыми признаками могут быть хейлит (трещины, сухость губ), рецидивирующие заеды (глубокие трещины в углах рта), срединная трещина нижней губы, а также красная сухая кожа верхних век. Темные круги вокруг глаз, бледность кожи лица с землистым оттенком могут быть важными индикаторами атопической предрасположенности.
Как атопический дерматит влияет на психологию?Психоэмоциональное влияние не ограничивается случайными и кратковременными переживаниями из-за обострения сыпи. Неконтролируемые проявления атопического дерматита постоянно осложняют жизнь «атопику». Зуд вызывает дискомфорт, а иногда и приводит к бессоннице, раздражительности, депрессии и повышенной утомляемости. Сыпь на открытых участках тела приходится скрывать от окружающих, нужно все время помнить о том, что чего-то делать нельзя, или это снова вызовет зуд и сыпь.
Атопический дерматит оказывает сильное влияние на весь жизненный уклад и мировоззрение человека, иногда делает его действительно «иным», по-другому смотрящим на мир, в котором так много потенциально опасных врагов-аллергенов, которые нормально переносят остальные. Считается, что «атопики» более склонны к интеллектуальным занятиям, вдумчивому и осторожному анализу происходящего вокруг, более чувствительны и замкнуты.
Проявления атопического дерматита оказывают влияние не только на психологию пациента, но и на всю семью, окружающих его людей и их взаимные отношения.
Что делать атопику? Нас, «атопиков», много: 1-3% всех взрослых европейцев и 10-20% школьников. Вот как получается: «дело-то житейское!» Мы сможем преодолеть и стресс и атопический дерматит.
Нас, «атопиков», много: 1-3% всех взрослых европейцев и 10-20% школьников. Вот как получается: «дело-то житейское!» Мы сможем преодолеть и стресс и атопический дерматит.
Чтобы победить эмоциональный стресс и социальные проблемы, нужна уверенность в себе и возможности контролировать состояние своей кожи. Нужно открытое, доверительное обсуждение своего состояния с друзьями, родственниками, врачами, другими людьми с такой же проблемой.
Для поддержания состояния ремиссии требуется специальный уход за кожей, дерматологами разработаны специальные средства — эмоленты. Многие известные марки косметики имеют линейку средств для атопической
кожи: Авен, Ля Рош Позе и др. При постоянном использовании гелей для мытья и кремов для кожи тела из этой серии, мы восстанавливаем защитный барьер кожи и устраняем неадекватные реакции.
Что еще можно использовать самостоятельно при атопии? Конечно же, сорбенты! Это вещества, которые принимаются внутрь, связывают токсины, различные соединения, и выводят их из организма естественным путем. как один из сорбентов знаю давно. Сорбенты показаны при лечении атопического дерматита. Маленьким пациентам «атопикам» трудно залпом выпить стакан воды с таблетками-сорбентами. А мелкий порошок наиболее подходит для приема детьми, легко разводится в малом количестве воды, в отличие от крупнозернистых аналогов, что облегчает его прием. Затем уже можно в течение получаса выпить стакан воды. После двухнедельного курса, часто признаки начавшегося обострения проходят, даже без использования специфического лечения. Взрослыми атопиками сорбент принимается как во время острой фазы дерматита, так и для профилактики перед сезонным обострением.
Где должен лечиться пациент с атопическим дерматитом?Необходимо наблюдаться и лечиться у одного и того же врача дерматолога. Если необходимо, дерматолог назначает консультации врачей-специалистов: невролога, гастроэнтеролога, аллерголога, вертебролога и др. Только так можно обеспечить преемственность различных видов терапии, проследить реакцию на них пациента.
К сожалению, приходится сталкиваться с пациентами, которые, не пройдя до конца назначенный курс лечения, ищут новомодные наружные средства, ходят от одного врача к другому, и каждый раз, не получив ожидаемого эффекта, переживают дополнительный стресс, за которым следует обострение заболевания. Не следует заниматься самолечением. Это серьезное заболевание, которое может приводить к опасным осложнениям, и, как было сказано, к развитию других форм «атопии». Для контроля над болезнью, профилактики осложнений необходимо следовать рекомендациям лечащего врача дерматолога.
Самые редкие болезни в мире
И примитивные пещерные люди, и современные блестящие ученые — человечество всегда боролось и борется со множеством болезней. Есть группа наследственных заболеваний, которых трудно избежать, если ваши родители ими болели; некоторые недуги — продукт непредсказуемых генетических мутаций. Большая группа заболеваний — результат размножения и процветания микроскопических организмов, поселившихся внутри человеческого тела.
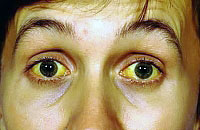
Синдром Жильбера
![]() Синдром Жильбера (СЖ) относится к генетическим заболеваниям и приводит к нарушению обмена билирубина, что может провоцировать доброкачественную неконъюгированную гипербилирубинемию. Обмен билирубина нарушается вследствие недостатка глюкуронилтрансферазы — особого печеночного фермента. Это и провоцирует рост уровня несвязанного билирубина в крови и возникновение желтухи. В синдром Жильбера, как правило, не несет угрозы плоду. Наличие СЖ увеличивает риск развития желчнокаменной болезни.
Синдром Жильбера (СЖ) относится к генетическим заболеваниям и приводит к нарушению обмена билирубина, что может провоцировать доброкачественную неконъюгированную гипербилирубинемию. Обмен билирубина нарушается вследствие недостатка глюкуронилтрансферазы — особого печеночного фермента. Это и провоцирует рост уровня несвязанного билирубина в крови и возникновение желтухи. В синдром Жильбера, как правило, не несет угрозы плоду. Наличие СЖ увеличивает риск развития желчнокаменной болезни.
Симптомы синдрома Жильбера проявляются более ярко под воздействием стрессов, физических нагрузок, недоедания, после вирусных заболеваний и вследствие употребления алкоголя и некоторых лекарственных препаратов. Для болезни Жильбера характерны:
- астения;
- пожелтение склер и слизистых разной степени (желтушность кожи наблюдается не всегда);
- боли в области печени;
- повышенный уровень билирубина в крови;
- нарушение работы желудка и болезненное пищеварение.
Синдром Жильбера бывает врожденным, в таком случае симптомы проявляются в возрасте от 12 до 30 лет. Второй вид синдрома — постгепатитная гипербилирубинемия, проявляющаяся после перенесенного вирусного гепатита. Во втором случае необходимо провести дифференциальную диагностику, чтобы не спутать СЖ с хроническим гепатитом.
Диагностика синдрома Жильбера
Для проведения диагностических исследований и планирования лечебных мероприятий необходимо обратиться к терапевту, генетику, гематологу и гастроэнтерологу (гепатологу). При подозрении на синдром Жильбера, помимо сбора анамнеза и физикального обследования, назначаются следующие методы диагностики:
- — при СЖ наблюдается повышение гемоглобина (>160 г/л), возможно появление ретикулеза и снижения осмотической стойкости эритроцитов.
- Биохимический анализ крови — билирубин может достигать 6 мг/дл, но в основном не переходит границу 3 мг/дл. Параметры, определяющие функцию печени, остаются в норме. Щелочная фосфатаза может повыситься.
- ПЦР — генетический маркер СЖ — количество ТА-повторов в промоторной цепи гена UGT1A1.
- УЗИ желчного пузыря и дуоденальное зондирование — почти у всех пациентов с СЖ наблюдаются изменения биохимического состава желчи.
- Биопсия печени — возможны патологические изменения в органе.
- Проба с голоданием — при наличии СЖ недоедание влечет за собой повышение билирубина в сыворотке крови.
- Проба с фенобарбиталом — применение фенобарбитала на фоне СЖ способствует понижению уровня неконъюгированного билирубина.
- Проба с никотиновой кислотой при СЖ провоцирует повышение содержания неконъюгированного билирубина. Такая же реакция происходит при введении рифампицина.
Врач может назначить дополнительные исследования и проведение дифференциальной диагностики СЖ с иными гипербилирубинемиями.
 Прогноз вполне благоприятный, в силу того что болезнь Жильбера относительно безопасна и специальное лечение не требуется (оно носит скорее бытовой характер). Основу терапии составляет соблюдение нормального режима питания, труда и отдыха. Во время обострений нужно соблюдать диету № 5 (отказ от жирной и жареной пищи, алкоголя), принимать витамины и желчегонные средства. Терапевт может назначить курс гепатопротекторов. Важно помнить, что не стоит прибегать к теплым физиопроцедурам. Лечение СЖ направлено на восстановление нормального уровня уридиндифосфат-глюкуронилтрансферазы (фермента печени) и стабилизацию общего самочувствия пациента.
Прогноз вполне благоприятный, в силу того что болезнь Жильбера относительно безопасна и специальное лечение не требуется (оно носит скорее бытовой характер). Основу терапии составляет соблюдение нормального режима питания, труда и отдыха. Во время обострений нужно соблюдать диету № 5 (отказ от жирной и жареной пищи, алкоголя), принимать витамины и желчегонные средства. Терапевт может назначить курс гепатопротекторов. Важно помнить, что не стоит прибегать к теплым физиопроцедурам. Лечение СЖ направлено на восстановление нормального уровня уридиндифосфат-глюкуронилтрансферазы (фермента печени) и стабилизацию общего самочувствия пациента.
Необходимо проконсультироваться с терапевтом и гепатологом, какие препараты можно применять на фоне синдрома Жильбера, а от каких придется отказаться (например, анаболические стероиды, глюкокортикоиды, кофеин и парацетамол могут усилить проявление желтухи).
Рак кишечника: симптомы и факторы риска
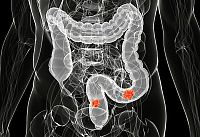 Рак кишечника — это злокачественное образование, которое формируется на слизистых оболочках в разных отделах кишечника. Чаще всего опухоль может развиться из полипов, однако далеко не все они превращаются в онкологию.
Рак кишечника — это злокачественное образование, которое формируется на слизистых оболочках в разных отделах кишечника. Чаще всего опухоль может развиться из полипов, однако далеко не все они превращаются в онкологию.
Рак кишечника по мере развития может нарушать нормальную работу органа и провоцировать кровотечения, в том числе заметные в каловых массах. При несвоевременной диагностике рака кишечника по симптомам он может распространиться на другие органы, что существенно ухудшит прогноз лечения.
Рак кишечника: факторы риска
Точных причин появления рака кишечника официальная медицина не называет, однако выделяет основные факторы риска, которые увеличивают вероятность развития онкологических опухолей в кишечнике:
- возраст: чаще всего раком кишечника болеют люди после 50 лет;
- нездоровый образ жизни: низкая физическая активность, вредная пища, лишний вес, злоупотребление алкоголем и курение;
- : воспалительные процессы в кишечнике (болезнь Крона, язвенный колит и пр.) считаются предраковыми заболеваниями, провоцирующими развитие онкологии;
- наследственность: риск возникновения рака повышается, если ближайшие родственники страдали им или другими кишечными заболеваниями.
Рак кишечника: симптомы
 На начальных стадиях рак кишечника симптомами может и не обратить на себя внимание пациента. Изменение частоты испражнений (в сторону уменьшения или учащения), беспричинное снижение веса, утомляемость и слабость, анемия, боли в заднем проходе — вот основные признаки, которыми характеризуется начало онкологического процесса в кишечнике.
На начальных стадиях рак кишечника симптомами может и не обратить на себя внимание пациента. Изменение частоты испражнений (в сторону уменьшения или учащения), беспричинное снижение веса, утомляемость и слабость, анемия, боли в заднем проходе — вот основные признаки, которыми характеризуется начало онкологического процесса в кишечнике.
С развитием заболевания может возникнуть , постоянные боли в животе, вздутие и ухудшение общего самочувствия. Возникновение любого из симптомов должно стать веским поводом для незамедлительного посещения врача.
При выявлении рака на ранних стадиях прогноз лечения достаточно оптимистичный — около 90 % пациентов остаются живы и больше не обращаются с жалобами к врачу. Однако большинство людей пропускают первые симптомы, принимая их за расстройство или геморроидальные процессы, запускают заболевание, и тогда даже хирургическое вмешательство позволяет сохранить жизнь только примерно 60 % пациентов.
Зачем нужны лишние зубы?
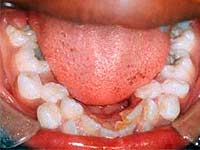 Сверхкомплектные зубы, или гипердонтия — одна из наследственно детерминированных аномалий количества зубов, довольно распространенных в настоящее время. Около 2-3% больных с пороками развития зубочелюстной системы имеют , дополнительно к 20 молочным или 32 постоянным единицам зубного ряда. Природа этой патологии до конца не ясна, считается, что ее возникновение связано с нарушением закладки зубов, а точнее, с нарушением механизма расщепления зубной пластинки, в результате чего формируется большее, чем положено количество зубных зачатков.
Сверхкомплектные зубы, или гипердонтия — одна из наследственно детерминированных аномалий количества зубов, довольно распространенных в настоящее время. Около 2-3% больных с пороками развития зубочелюстной системы имеют , дополнительно к 20 молочным или 32 постоянным единицам зубного ряда. Природа этой патологии до конца не ясна, считается, что ее возникновение связано с нарушением закладки зубов, а точнее, с нарушением механизма расщепления зубной пластинки, в результате чего формируется большее, чем положено количество зубных зачатков.
Где искать лишние зубы?
Сверхкомплектные зубы могут обнаруживаться в детском возрасте на молочном прикусе, но чаще они выявляются после смены зубов в постоянном прикусе.
Обычно лишние зубы появляются около средних верхних резцов, моляров, премоляров, клыков, реже между нижних резцов, премоляров и клыков. Они могут вырастать на зубной дуге, а могут располагаться в преддверии полости рта или непосредственно в полости рта в области верхнего неба.
По форме сверхкомплектные зубы могут быть похожи на обычные постоянные единицы зубного ряда, но чаще имеют каплевидную или шиповидную форму. Они могут располагаться обособленно, спаиваться с постоянными зубами, образовывать целые зубные конгломераты и зубоподобные образования.
Иногда сверхкомплектные зубы «скрыты» от глаз, то есть ретинированы, тогда они обнаруживаются только при рентгенологическом исследовании.
Чем опасны сверхкомплектные зубы?
Сверхкомплектные единицы вмешиваются в формирование зубных рядов и затрудняют прорезывание постоянных зубов. При значительных размерах челюсти лишний зуб не нарушает структуру зубного ряда, а при небольшой челюсти непременно становится причиной аномалий положения комплектных зубов, что имеет негативные эстетические, функциональные и нередко психологические последствия для человека.
У детей с аномалиями развития зубов часто снижен аппетит, они медленнее пережевывают пищу, нередко у них нарушено глотание, все это становится причиной развития заболеваний пищеварительной системы.
Тесное расположение зубов, их неправильное положение затрудняют процессы самоочищения зубного ряда и проведение гигиенических процедур. Это создает благоприятные условия для развития кариеса, гингивита, периодонтита и пародонтоза, приводящих к разрушению твердых тканей зубов и их выпадению.
Обладатели лишних зубов часто шепелявят. Расстройства речи и косметические недостатки становятся причиной насмешек над ребенком, формируют из него малообщительную и замкнутую личность, нередко сказываются на его психическом развитии.
Диагностика гипердонтии
В большинстве случаев сверхкомплектные зубы обнаруживаются во время прорезывания передних зубов. Чтобы уточнить их количество и месторасположение, необходима рентгенодиагностика, однако простой рентгенографией не обойтись, поскольку тени от лишних зубов накладываются на контуры комплектных элементов зубного ряда.
Для точной диагностики гипердонтии используют рентгенографию внутри рта со снимками в различных проекциях. При множественной ретенции сверхкомплектных зубов полезную информацию о взаимном расположении сверхкомплектных и постоянных зубов дает ортопантомография.
Что делать со сверхкомплектными зубами?
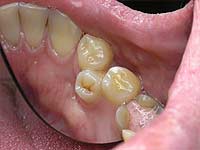 Подход к лечению гипердонтии дифференцированный, терапия зависит от места расположения лишних зубов. В целом сверхкомплектные зубы подлежат удалению как можно раньше, особенно, если они нарушают анатомию зубного ряда, вызывают болезненные переживания у больного. При ликвидации лишних зубов в детском возрасте нормальная форма зубного ряда нередко восстанавливается благодаря механизмам саморегуляции организма, но если упустить время, без последующего ортодонтического лечения не обойтись.
Подход к лечению гипердонтии дифференцированный, терапия зависит от места расположения лишних зубов. В целом сверхкомплектные зубы подлежат удалению как можно раньше, особенно, если они нарушают анатомию зубного ряда, вызывают болезненные переживания у больного. При ликвидации лишних зубов в детском возрасте нормальная форма зубного ряда нередко восстанавливается благодаря механизмам саморегуляции организма, но если упустить время, без последующего ортодонтического лечения не обойтись.
Иногда в целях сохранения функций зубного ряда, наоборот, жертвуют дистопированным постоянным зубом, сохраняя при этом выгодно расположенный, анатомически полноценный сверхкомплектный зуб.
Если сверхкомплектный зуб вырастает на месте непрорезавшегося постоянного, вначале определяют степень его полноценности. В случае если сверхкомплектный зуб устойчив, имеет развитый корень и более или менее анатомически правильную форму, а непрорезавшийся «законный владелец места» при этом несостоятелен и бесперспективен, предпочтение отдают «захватчику».
Удаление ретинированных, погруженных в ткани челюсти сверхкомплектных зубов, представляет определенные трудности, связанные с глубиной их залегания, неправильной формой, близостью к корням и зачаткам постоянных зубов. Однако рациональный оперативный подход к сверхкомплектному зубу с учетом результатов рентгенологического исследования, позволяет успешно решать эти проблемы.
У детей сверхкомплектные зубы удаляют под общим или местным обезболиваем, на фоне действия успокаивающих препаратов, у взрослых — достаточно местной анестезии.
Порфирия - научно-обоснованный «вампиризм»
Вампиризм — современная субкультура, объединяющая молодежь, которая считает себя вампирами. В основном интерес ограничивается изучением вампирской тематики в искусстве и имитацией внешнего вида любимых персонажей, молодые люди вряд ли задумываются над историей происхождения образов вампиров.
Случаи «ненаучного» вампиризма
Движение «вампиризм» зародилось в 1970 году благодаря поклонникам творчества Энн Райс, автора знаменитого романа «Интервью с вампиром». Вместе с тем, вампирская тематика своими корнями уходит в далекое прошлое и отражена в фольклоре многих народов.
Образ вампира в искусстве складывался годами, но ярче всего отражен в опубликованном в 1897 г. готическом романе ирландского писателя Брэма Стокера «Дракула», последующие произведения обязаны своим существованием именно этому бессмертному творению.
Каковы литературные признаки вампиризма?
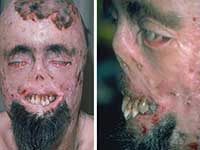 Чаще всего вампиры изображаются как интеллигентные, элегантные, загадочные и сексуальные особы, ведущие замкнутый образ жизни.
Чаще всего вампиры изображаются как интеллигентные, элегантные, загадочные и сексуальные особы, ведущие замкнутый образ жизни.
Кровь нужна им для того, чтобы поддерживать обмен веществ и не умереть. Согласно легендам:
- вампиры боятся солнечного света, защищены темной одеждой, выходят только под покровом ночи и возвращаются до рассвета;
- дневной свет убивает вампира и снижает его силы;
- они избегают званых обедов и ужинов, им чужда человеческая пища;
- они бледны, кожа тонкая и ранимая, холодная на ощупь;
- неизменный — клыки и тронутые пурпуром десны;
- глаза вампира окружены дымкой пушистых ресниц, белки красноваты, а зрачки затуманены;
- им свойственна тревожность, мнительность, агрессивность, впадают в неистовство в момент острого желания крови, могут превращаться в монстров в физическом и психологическом смысле.
порфирия
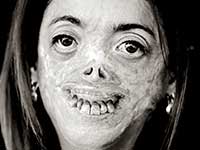 Не владея знаниями в области медицины, черпая вдохновение в ирландских мифах о вампирах, в преданиях народов Трансильвании, исторических описаниях жизни Влада Цепеша, прообраза Дракулы, Стокер, сам того не подозревая, описал страдания тяжело больного порфирией человека.
Не владея знаниями в области медицины, черпая вдохновение в ирландских мифах о вампирах, в преданиях народов Трансильвании, исторических описаниях жизни Влада Цепеша, прообраза Дракулы, Стокер, сам того не подозревая, описал страдания тяжело больного порфирией человека.
Порфирия, иначе — пурпурная болезнь, группа заболеваний, связанных с нарушением обмена порфирина, ярко- красного пигмента. В основе патологии лежат нарушения синтеза гема — соединения порфирина с железом, основа эритроцитов крови человека. Сбой в системе образования гема приводит к анемии, накоплению в организме продуктов промежуточного обмена, оказывающих токсическое действие на органы и системы, обусловливающие типичную симптоматику «вампирской» болезни.
Причины развития порфирии лежат на генетическом уровне, заболевание передаются по наследству . Вероятность передачи гена порфирии довольно высока, больной родитель «дарит» дефектный ген ребенку в 50% случаев, независимо от пола, однако только в 20% случаев разворачивается клиническая картина заболевания. Для его манифестации необходимо действие провоцирующих факторов : некоторых лекарственных препаратов, инфекции, гормональных перестроек, определенной пищи и алкоголя — не зря мифические вампиры сторонились человеческих застолий.
Симптомы порфирии
Как болезни чаще встречаются среди мужчин и проявляются в весенне-летние месяцы. Типичным признаком порфирии является моча красно—бурого цвета , что обусловлено наличием недоокисленного порфириногена, превращающегося на свету в пурпурный порфирин.
Острая порфирия проявляется сильными болями в животе, пояснице, в конечностях, тахикардией, повышением артериального давления, рвотой, мышечной слабостью, психомоторным возбуждением, галлюцинациями, бредом, эпилептиформными припадками и другими симптомами, развивающимися в результате острого отравления организма, образующимися продуктами обмена порфирина и диффузного поражения периферической и центральной нервной системы.
Порфирия имеет весьма типичные внешние проявления.
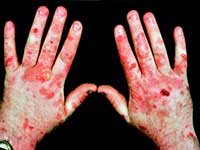
Можно себе представить реакцию людей в средние века, когда они сталкивались с больным порфирией. Подобное зрелище оставляло след в памяти, запечатлевалось в легендах, обрастало мифическими подробностями.
Порфирия тяжелое заболевание, лечится в основном инфузией гема. Дополнительно назначаются симптоматические средства, плазмоферез, кровь необходима больным для того, чтобы справляться с интоксикацией и чтобы просто жить.
Настоящая проблема для мужчины: бесплодие и его причины
 Говорят, каждый мужчина в своей жизни должен посадить дерево, построить дом и вырастить сына. И если с первыми двумя задачами, при желании, справляется практически каждый представитель сильного пола, то, к сожалению, при решении последней из них, до 8 % мужчин на Земле сталкиваются с серьезными проблемами. И эта цифра не учитывает тех, кто смирился с бесплодием и предпочел не делиться своей проблемой с врачами.
Говорят, каждый мужчина в своей жизни должен посадить дерево, построить дом и вырастить сына. И если с первыми двумя задачами, при желании, справляется практически каждый представитель сильного пола, то, к сожалению, при решении последней из них, до 8 % мужчин на Земле сталкиваются с серьезными проблемами. И эта цифра не учитывает тех, кто смирился с бесплодием и предпочел не делиться своей проблемой с врачами.
Бесплодие у мужчины — причина бездетности пары
Бесплодие — не просто отсутствие детей, оно диагностируется в том случае, если пара, активно живущая половой жизнью без использования средств контрацепции, в течение года испытывает трудности с зачатием. Немного уточним понятие «активная половая жизнь», для одного это ежедневный секс, для другого — пару раз в месяц. В случае с бесплодием — «активная» означает не менее 1 раза в неделю.
По статистике в 40% случаев отсутствия детей у пары виновно именно . Причины его могут быть различными, на первый взгляд безобидными и легко преодолимыми, или настолько весомыми, что устранить их невозможно.
Мужское бесплодие, как собственно и женское, может быть абсолютным и относительным. С абсолютным бесплодием мужчине придется смириться, если у него удалены семенные железы. Другие варианты бесплодия являются потенциально относительными, требуют углубленной диагностики для поиска причины и решения проблемы.
Варианты мужского бесплодия: причины и формы
 Процесс формирования полноценных сперматозоидов довольно сложен и контролируется гормонопродуцирующей частью головного мозга — гипофизарно-гипоталамической системой. Половые клетки, которые образуются в яичке, дозревают по пути к семенным пузырькам, но отсутствие проблем в этой части производства, не гарантирует успешного оплодотворения. На репродуктивную способность мужчины оказывает влияние состояние всех мужских органов и всего организма в целом.
Процесс формирования полноценных сперматозоидов довольно сложен и контролируется гормонопродуцирующей частью головного мозга — гипофизарно-гипоталамической системой. Половые клетки, которые образуются в яичке, дозревают по пути к семенным пузырькам, но отсутствие проблем в этой части производства, не гарантирует успешного оплодотворения. На репродуктивную способность мужчины оказывает влияние состояние всех мужских органов и всего организма в целом.
Что вызывает этой проблемы?
- Гипоталамо-гипофизарные нарушения, возникающие у головном мозге, на уровне гормональной регуляции сперматогенеза.
- Тестикулярные, связанные с яичками.
- Посттестикулярные, возникающие по ходу движения спермиев от яичка до выхода из уретры.
- Иммунологические с наличием антиспермальных антител, убивающих сперматозоиды.
- Эякуляторные расстройства, то есть проблема семяизвержения.
- Сексуальные расстройства, нарушающие «доставку» спермы в половые органы женщины, такие как эректильная дисфункция и сниженное половое влечение.
На способность мужчины к продолжению рода негативно влияют:
- неблагоприятная экологическая обстановка;
- стресс;
- прием лекарственных препаратов в высоких дозах;
- курение;
- прием алкоголя;
- употребление наркотиков.
Тестикулярное бесплодие
Тестикулярное бесплодие возникает при заболеваниях яичка и его инертности.
- Варикозное расширение вен мошонки и семенного канатика (варикоцеле).
- Крипторхизм, двусторонний не устраненный или устраненный довольно поздно, когда семяпродуцирующая ткань яичек уже атрофировалась.
- Перекрут яичка, приводящий к резкому нарушению его функции.
- Орхит или воспаление яичка, в результате которого умирают клетки-предшественники сперматозоидов.
- Гипергонадотропный гипогонадизм — недоразвитие яичек.
- Генетические причины, вызывающие нарушение формирования половой системы и образования яичек.
- Резистентность к андрогенам, когда сперматогенный эпителий яичек остается глух к гормональным сигналам, провозглашающим образование половых клеток.
- Необструктивная азооспермия, состояние, при котором продукция спермиев не нарушена, движение их по семявыносящим путям не затруднено, но на выходе не обнаруживается «ни одного» живого экземпляра, способного продолжить род.
Посттестикулярные причины бесплодия
Эти причины приводят к нарушению созревания сперматозоидов, их гибели, ослаблению их оплодотворяющей способности, а также препятствует передвижению спермиев по семявыносящим протокам.
- Инфекции половых органов, особенно протекающие длительно и скрыто, например, хламидиоз, трихомониаз, микоплазмоз, уреаплазмоз, цитомегаловирусная инфекция, герпес.
- Неспецифические воспаления простаты, уретры и семенных пузырьков.
- Отсутствие придатка яичка, в котором «дозревают» сперматозоиды, и протока, по которому они выходят из яичка.
- Закупорка семявыносящих протоков или их удаление.
Наконец, существуют варианты, когда причины снижения репродуктивной функции установить не удается, так называемое бесплодие идиопатическое. В любом случае, чтобы получить окончательный диагноз, необходимо обратиться у урологу и пройти полное обследование.
Сахарный диабет I типа, а именно он обнаруживается у детей, относится к заболеваниям, предрасположенность к которым передается по наследству. Именно предрасположенность, но не сам недуг, который развивается только тогда, когда на организм малыша действуют провоцирующие внешние и внутренние факторы. Все причины сахарного диабета не выявлены до сих пор, но считается, что пусковым моментом заболевания чаще всего становится инфекция или стресс.

Казалось бы, если родители или другие родственники ребенка осведомлены о причинах и признаках сахарного диабета, поскольку сами им страдают, никаких трудностей в раннем выявлении недуга у ребенка быть не должно. Однако, к сожалению, чаще всего заболевание обнаруживается именно в
поздней стадии, когда малыш уже находится в тяжелом состоянии и жизненно нуждается в интенсивных медицинских мероприятиях. Как ни грустно говорить, но на этой стадии сахарный диабет очень трудно поддается лечению и вызывает появление тяжелых осложнений. Ситуация осложняется тем, что детский диабет имеет очень короткий скрытый период, а значит, времени на длительные раздумья нет.
Каждый взрослый член семьи должен четко знать первые признаки сахарного диабета у детей и уметь трезво оценивать те показатели здоровья, которые наиболее важны с точки зрения ранней диагностики болезни.
Какие признаки сахарного диабета должны заметить родители?
- Изменение аппетита:
- появление неестественной для ребенка тяги к сладкому;
- тремление есть часто, то есть ребенок из-за сильного чувства голода с трудом выдерживает традиционные 3-4 часовые перерывы между приемами пищи;
- слабость и сонливость через 1,5-2 часа после еды.
Конечно, большинство детей любит сладости, а многих клонит в сон после еды, но в случае наличия генетической предрасположенности к сахарному диабету I типа эти привычки могут быть первыми признаками болезни, причем их изолированное существование может свидетельствовать о том, что заболевание еще не зашло далеко, а значит, лечить его будет легче. - Ребенок худеет, несмотря на нормальный и даже повышенный аппетит.
Не нужно списывать снижение веса на то, что ребенок быстро растет, лучше обследовать его и убедиться, что изменение массы тела вызвано повышением физической активности и ростом потребностей малыша, а не тем, что его организм из последних сил пытается бороться с заболеванием. Несмотря на то, что в крови содержится избыток глюкозы, клетки организма ребенка испытывают жесточайший голод, ведь поджелудочная железа при сахарном диабете практически не синтезирует инсулин, необходимый для того, чтобы глюкоза была усвоена тканями. - Ребенок быстро устает, становится вялым, сонным.
При отсутствии повышенной температуры, кашля и других признаков простуды — эти симптомы должны насторожить с точки зрения сахарного диабета. - Ребенок начинает больше пить и чаще мочиться, хотя этот симптом признается диабетологами поздним.
Как и соль, сахар притягивает к себе жидкость, организм, пытаясь «разбавить» сахар, требует поступления воды, сигнализируя об этом жаждой. Рано или поздно при сахарном диабете почки перестают удерживать глюкозу в организме, она начинает выделяться с мочой, что в свою очередь ведет к увеличению диуреза. Дети больные сахарным диабетом начинают вставать ночью в туалет, а иногда мочиться в постель. - Пятна мочи на горшке, в туалете, на подгузниках становятся липкими.
Это чисто физическое явление, раствор сахара по понятным причинам после испарения воды оставляет после себя липкие пятна. Внимательная мама всегда заметит этот симптом.
Тошнота , рвота, боли в животе, сухость и зуд кожи, признаки нейродермита, упорный фурункулез, пиодермия, нарушение зрения — это поздние симптомы и последствия сахарного диабета, признак того, что болезнь уже набрала силу и приостановить ее развитие будет крайне сложно. А ведь обратись родители к эндокринологу при появлении самых первых сигналов сахарного диабета, заболевание можно было бы выявить на ранних этапах, даже еще до того, как нарушится работа поджелудочной железы и повысится уровень сахара в крови. Время не было бы упущено и врачам удалось бы сохранить детские силы для борьбы с недугом и для полноценной жизни.
Внимание! О наличии сахарного диабета у ребенка должны быть осведомлены все взрослые люди: контактирующие с ним воспитатели, учителя, соседи, друзья. Во-первых, это позволит избежать ошибок в питании малыша, во-вторых, в случае внезапного ухудшения состояния здоровья доставить его в специализированное эндокринологическое отделение, а не, к примеру, в инфекционную больницу или хирургию.
Среди наследственных болезней, развивающихся в результате мутаций, традиционно выделяют три подгруппы:
- моногенные наследственные заболевания
- полигенные наследственные болезни
- хромосомные аберрации
От наследственных заболеваний следует отличать врождённые заболевания, которые обусловлены внутриутробными повреждениями, вызванными, например, инфекцией (сифилис или токсоплазмоз) или воздействием иных повреждающих факторов на плод во время беременности.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое, порой весьма долгое, время.
Моногенные наследственные заболевания
Моногенные болезни наследуются в соответствии с законами классической генетики Менделя. Соответственно этому, для них генеалогическое исследование позволяет выявить один из трёх типов наследования: аутосомно-доминантный, аутосомно-рецессивный и сцепленное с полом наследование.
Это наиболее широкая группа наследственных заболеваний. В настоящее время описано более 4000 вариантов моногенных наследственных болезней, подавляющее большинство которых встречается довольно редко (например, частота серповидноклеточной анемии - 1/6000).
Широкий круг моногенных болезней образуют наследственные нарушения обмена веществ, возникновение которых связано с мутацией генов, контролирующий синтез ферментов и обусловливающих их дефицит или дефект строения - ферментопатии.
Полигенные наследственные болезни
Полигенные болезни наследуются сложно. Для них вопрос о наследовании не может быть решён на основании законов Менделя. Ранее такие наследственные заболевания характеризовались как болезни с наследственной предрасположенностью. К этим заболеваниям относятся такие болезни как рак, сахарный диабет, шизофрения, эпилепсия, ишемическая болезнь сердца, гипертензия и многие другие.
Хромосомные аберрации
Хромосомные болезни обусловлены грубым нарушением наследственного аппарата - изменением числа и структуры хромосом. Типичная причина, в частности, - алкогольная интоксикация родителей при зачатии («пьяные дети»). Сюда относятся синдромы Дауна, Клaйнфельтера, Шерешевского - Тернера, Эдвардса, «кошачьего крика» и другие.
Диагностика и лечение наследственных болезней
В последнее время складывается мнение, что относительно высокая частота наследственных заболеваний обусловлена определёнными преимуществами «мутантов» по отношению к факторам естественного отбора или с «предрасположенностью к болезни».
Терапия наследственных заболеваний включается в себя симптоматическое лечение и генотерапию.
Симптоматическое лечение
Наследственным заболеваниям свойственны различные симптоматические проявления, и их лечение во многом является симптоматическим. Отдельные нарушения обмена веществ исправляют назначением специальных диет, направленных на уменьшение токсических веществ в организме, накопление которых обусловлено мутациями в определённых генах. Например, при фенилкетонурии назначают безаланиновую диету.
Для ослабления симптомов наследственных болезней, связанных с дефектом определённого белка, вводят внутривенно такую его функциональную форму, которая не вызывает иммунной реакции. Такая замещающая терапия применяется при лечении гемофилии, тяжёлого комбинированного иммунодефицита и др. Иногда для компенсации определённых утраченных функций проводят трансплантацию костного мозга и других органов. Существующая терапия, к сожалению, в подавляющем большинстве случаев мало эффективна.
Генная терапия
Принципиально новым методом, эффективным и направленным на уничтожение генетической причины наследственного заболевания, является генотерапия. Суть метода генотерапии - введение нормальных генов в дефектные клетки.
Концепция генной терапии заключатся в том, что наиболее радикальным способом борьбы с разного рода заболеваниями, вызываемыми изменениями генетического содержания клеток, должна быть обработка, направленная непосредственно на исправление или уничтожение самой генетической причины заболевания, а не ее следствий.
В связи с тем, что генная терапия представляет собой новое направление медицинской генетики, а болезни, которые пытаются лечить этим способом, очень разнообразны, создано множество оригинальных подходов к этой проблеме. В настоящее время исследования по генотерапии в основном направлены на коррекцию генетических дефектов соматических, а не половых клеток, что связано с чисто техническими проблемами, а также из соображений безопасности.
По материалам статьи «Наследственные болезни»
 Поделки смешарики из СД дисков: Нюша, Бараш, Лосяш Поделки из цветной бумаги смешарики
Поделки смешарики из СД дисков: Нюша, Бараш, Лосяш Поделки из цветной бумаги смешарики Простая выкройка комбинезона: особенности построения чертежа Как выкроить модный комбинезон
Простая выкройка комбинезона: особенности построения чертежа Как выкроить модный комбинезон Необычные детские пледы крючком
Необычные детские пледы крючком